Part1 数据下载
先去Korean Personal Genome Project下载了编号为KPGP-00001的数据。
先说一下KPGP吧,中文名叫韩国个人基因组计划,这里面的数据都是可以免费下载的。


nohup wget -c -r -nd -np -k -L -p ftp://ftp.kobic.re.kr/pub/KPGP/2015_release_candidate/WGS/KPGP-00001 1>/dev/null 2>&1 &
简单的说一下这个命令中的几个知识点:
1.nohup
nohup命令可以在你退出帐户之后继续运行相应的进程。nohup就是不挂起的意思( no hang up)
2.&
当在前台运行某个作业时,终端被该作业占据;可以在命令后面加上& 实现后台运行
一般情况下,两者同时使用:
nohup command &
ps all 可以查所有后台任务
这样就不用管它了
下载完成后的数据量如下
一般大型的文件的下载需要将其md5文件一起下载,来检验下载的文件是否下载完全
md5sum KPGP-00001_L1_R1.fq.gz >md5tmp1.txt
cat md5tmp1.tx
cat KPGP-00001_L1_R1.fq.gz.md5
如果结果一致就可以判断下载完全
此时的目录应该如下:
Part2 测序reads的质控
cat fastqc.sh
#!/bin/bash
for i in $(seq 1 6)
do
fastqc /home/ubuntu/WGS_new/input/KPGP-00001_L${i}_R1.fq.gz /home/ubuntu/WGS_new/input/KPGP-00001_L${i}_R2.fq.gz -o /home/ubuntu/WGS_new/output/fastqc_result && echo "KPGP-00001_L${i} fasqtc is done" >fastqc${i}.log
done
nohup bash fastqc.sh &
由于测序数据有6个lane,而且又是pair-end测序,所以用到multiqc来归纳汇总。
cd ~/WGS_new/output/fastqc_result
multiqc *.zip
这一步之后产生的文件如下
├── input
│ ├── KPGP-00001_L1_R1.fq.gz
│ ├── KPGP-00001_L1_R2.fq.gz
│ ├── KPGP-00001_L2_R1.fq.gz
│ ├── KPGP-00001_L2_R2.fq.gz
│ ├── KPGP-00001_L3_R1.fq.gz
│ ├── KPGP-00001_L3_R2.fq.gz
│ ├── KPGP-00001_L4_R1.fq.gz
│ ├── KPGP-00001_L4_R2.fq.gz
│ ├── KPGP-00001_L5_R1.fq.gz
│ ├── KPGP-00001_L5_R2.fq.gz
│ ├── KPGP-00001_L6_R1.fq.gz
│ ├── KPGP-00001_L6_R2.fq.gz
│ └── nohup.out
├── output
│ └── fastqc_result
│ ├── KPGP-00001_L1_R1_fastqc.html
│ ├── KPGP-00001_L1_R1_fastqc.zip
│ ├── KPGP-00001_L2_R1_fastqc.html
│ ├── KPGP-00001_L2_R1_fastqc.zip
│ ├── KPGP-00001_L2_R2_fastqc.html
│ ├── KPGP-00001_L2_R2_fastqc.zip
│ ├── KPGP-00001_L3_R1_fastqc.html
│ ├── KPGP-00001_L3_R1_fastqc.zip
│ ├── KPGP-00001_L3_R2_fastqc.html
│ ├── KPGP-00001_L3_R2_fastqc.zip
│ ├── KPGP-00001_L4_R1_fastqc.html
│ ├── KPGP-00001_L4_R1_fastqc.zip
│ ├── KPGP-00001_L4_R2_fastqc.html
│ ├── KPGP-00001_L4_R2_fastqc.zip
│ ├── KPGP-00001_L5_R1_fastqc.html
│ ├── KPGP-00001_L5_R1_fastqc.zip
│ ├── KPGP-00001_L5_R2_fastqc.html
│ ├── KPGP-00001_L5_R2_fastqc.zip
│ ├── KPGP-00001_L6_R1_fastqc.html
│ ├── KPGP-00001_L6_R1_fastqc.zip
│ ├── KPGP-00001_L6_R2_fastqc.html
│ ├── KPGP-00001_L6_R2_fastqc.zip
│ ├── multiqc_data
│ │ ├── multiqc_data.json
│ │ ├── multiqc_fastqc.txt
│ │ ├── multiqc_general_stats.txt
│ │ ├── multiqc.log
│ │ └── multiqc_sources.txt
│ └── multiqc_report.html
└── script
├── fastqc1.log
├── fastqc2.log
├── fastqc3.log
├── fastqc4.log
├── fastqc5.log
├── fastqc6.log
├── fastqc.sh
└── nohup.out
此处的质量控制指的是原始数据的质量控制,接下来了解一下质控相关的知识
(此处有借鉴微信公众号:解螺旋的矿工的文章)
说到质量控制,先了解什么是质量值
碱基质量值就是能够用来定量描述碱基好坏程度的一个数值。它该如何才能恰当地描述这个结果呢?我们试想一下,如果测序测得越准确,这个碱基的质量就应该越高;反之,测得越不准确,质量值就应该越低。也就是说可以利用碱基被测错的概率来描述它的质量值,错误率越低,质量值就越高。
假定碱基的测序错误率为p_error,质量值为Q
有公式:
Q = -10log(p_error)
如果该碱基的测序错误率是0.01,那么质量值就是20(俗称Q20),如果是0.001,那么质量值就是30(俗称Q30)。Q20和Q30的比例常常被我们用来评价某次测序结果的好坏,比例越高就越好。
现在一般都是使用Phred33这个体系。
基本上可以从下面几个方面来了解原始数据的质量值:
1.reads各位置的碱基质量分布
在做read质量值分析的时候,FastQC并不单独查看具体某一条read中碱基的质量值,而是将Fastq文件中所有的read数据都综合起来一起分析。如下图,来自KPGP-00001_L1_R1
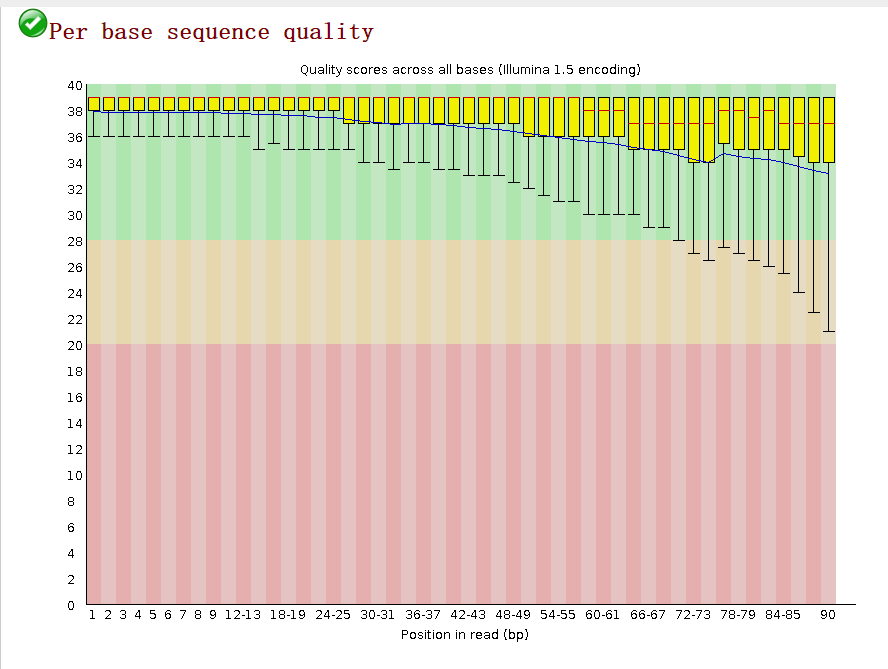
这个图的横轴是read上碱基的位置,纵轴是碱基质量值。
我们可以看到read上的每一个位置都有一个黄色的箱型图表示在该位置上所有碱基的质量分布情况。
2.碱基总体质量值分布
fastqc还会提供一个碱基总体的质量分布如下:
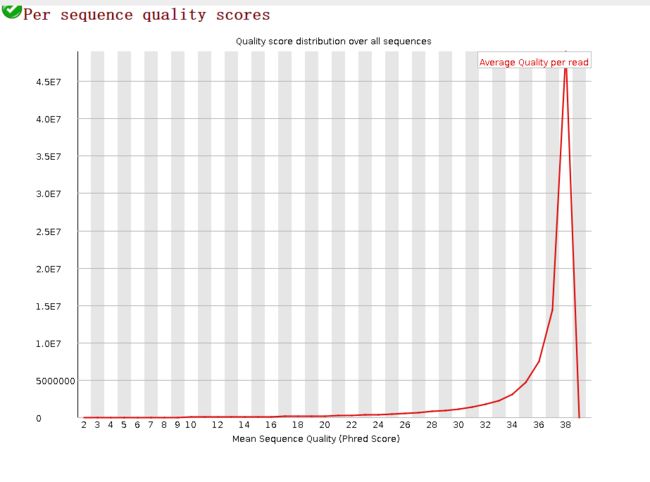
只要大部分都高于20,那么就比较正常
另外,Q20和Q30的比例是我们衡量测序质量的一个重要指标。一般来说,对于二代测序,最好是达到Q20的碱基要在95%以上(最差不低于90%),Q30要求大于85%(最差也不要低于80%)
3.reads各个位置上碱基比例分布
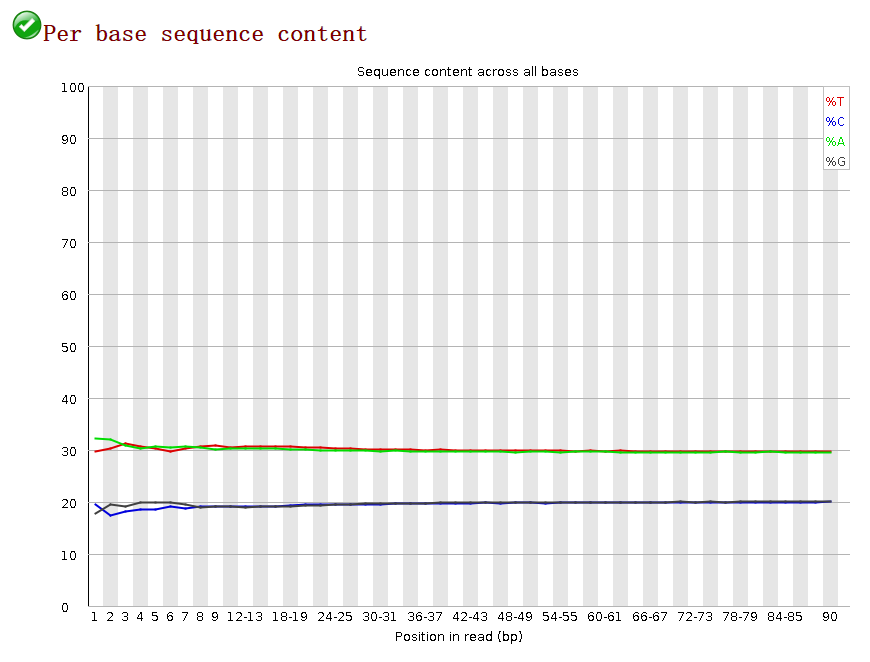
这个是为了分析碱基的分离程度。何为碱基分离?我们知道AT配对,CG配对,假如测序过程是比较随机的话(随机意味着好),那么在每个位置上A和T比例应该差不多,C和G的比例也应该差不多,如上图所示,两者之间即使有偏差也不应该太大,最好平均在1%以内,如果过高,除非有合理的原因,比如某些特定的捕获测序所致。
4.GC含量分布图

GC含量指的是G和C这两中碱基占总碱基的比例。二代测序平台或多或少都存在一定的测序偏向性,我们可以通过查看这个值来协助判断测序过程是否足够随机。对于人类来说,我们基因组的GC含量一般在40%左右。因此,如果发现GC含量的图谱明显偏离这个值那么说明测序过程存在较高的序列偏向性,结果就是基因组中某些特定区域被反复测序的几率高于平均水平,除了覆盖度会有偏离之后,将会影响下游的变异检测和CNV分析。图中的GC含量约在39%,处于正常范围。
5.N含量分布图
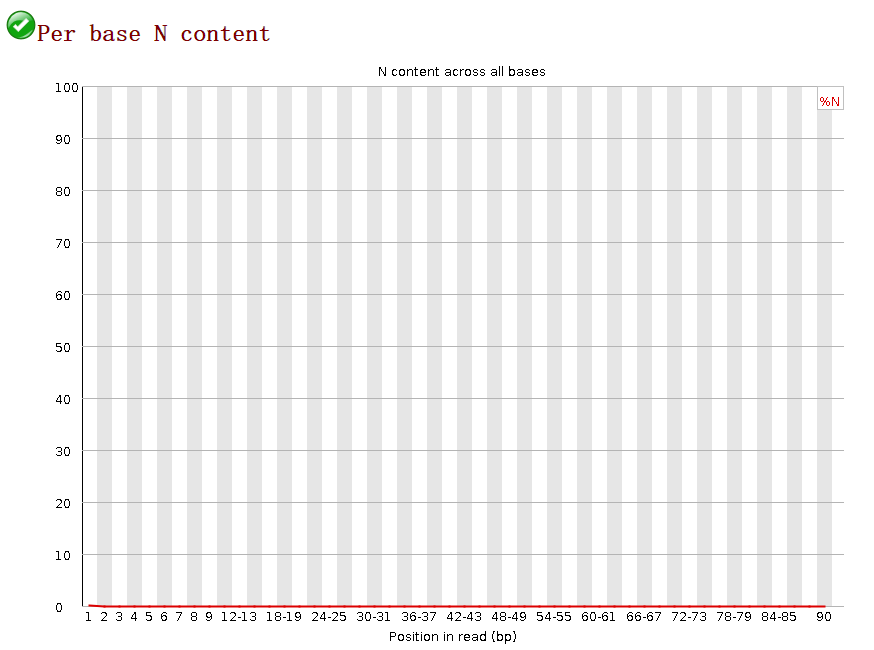
N在测序数据中一般是不应该出现的,如果出现则意味着,测序的光学信号无法被清晰分辨,如果这种情况多的话,往往意味着测序系统或者测序试剂的错误。图中接近0%。
6.接头序列
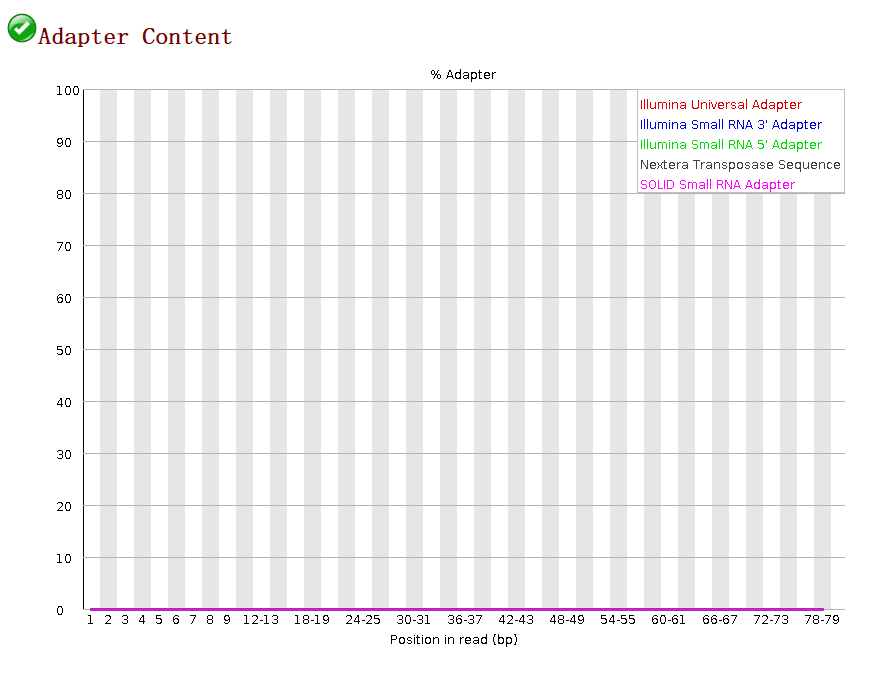
在测序之前需要构建测序文库,测序接头就是在这个时候加上的,其目的一方面是为了能够结合到flowcell上,另一方面是当有多个样本同时测序的时候能够利用接头信息进行区分。当测序read的长度大于被测序的DNA片段时,就会在read的末尾测到这些接头序列。一般的WGS测序是不会测到这些接头序列的,因为构建WGS测序的文库序列(插入片段)都比较长,约几百bp,而read的测序长度都在100bp-150bp这个范围。不过在进行一些RNA测序的时候,由于它们的序列本来就比较短很多只有几十bp长(特别是miRNA),那么就很容易会出现read测通的现象,这个时候就会在read的末尾测到这些接头序列。
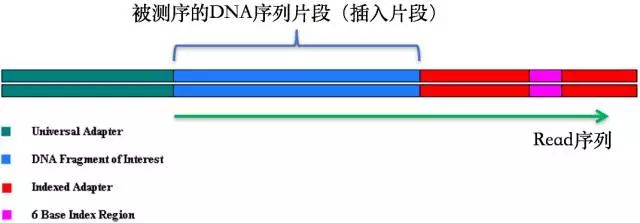
最后,如果测序质量很差的情况下,则需要切除测序接头序列和read的低质量序列
工具有:SOAPnuke、cutadapt、untrimmed等,但是,常用的一般是SOAPnuke、cutadapt、untrimmed等。
Part3 比对(mapping)
step1
mkdir /home/ubuntu/WGS_new/input/genome
cd /home/ubuntu/WGS_new/input/genome
wget http://hgdownload.cse.ucsc.edu/goldenPath/hg38/bigZips/hg38.fa.gz
#对下载的基因组建立索引,下面是建索引的脚本
#!/bin/bash
bwa index -a bwtsw -p /home/ubuntu/WGS_new/input/genome/hg38 /home/ubuntu/WGS_new/input/genome/Homo_sapiens_assembly38.fasta && echo "bwa index of hg38 is done"
#运行脚本
nohup bash bwa_index.sh &
索引构建完毕后的目录如下:
├── input
│ ├── genome
│ │ ├── hg38.amb
│ │ ├── hg38.ann
│ │ ├── hg38.bwt
│ │ ├── hg38.fa
│ │ ├── hg38.pac
│ │ ├── hg38.sa
│ │ └── nohup.out
│ ├── KPGP-00001_L1_R1.fq.gz
│ ├── KPGP-00001_L1_R2.fq.gz
│ ├── KPGP-00001_L2_R1.fq.gz
│ ├── KPGP-00001_L2_R2.fq.gz
│ ├── KPGP-00001_L3_R1.fq.gz
│ ├── KPGP-00001_L3_R2.fq.gz
│ ├── KPGP-00001_L4_R1.fq.gz
│ ├── KPGP-00001_L4_R2.fq.gz
│ ├── KPGP-00001_L5_R1.fq.gz
│ ├── KPGP-00001_L5_R2.fq.gz
│ ├── KPGP-00001_L6_R1.fq.gz
│ ├── KPGP-00001_L6_R2.fq.gz
│ └── nohup.out
├── output
│ └── fastqc_result
│ ├── KPGP-00001_L1_R1_fastqc.html
│ ├── KPGP-00001_L1_R1_fastqc.zip
│ ├── KPGP-00001_L2_R1_fastqc.html
│ ├── KPGP-00001_L2_R1_fastqc.zip
│ ├── KPGP-00001_L2_R2_fastqc.html
│ ├── KPGP-00001_L2_R2_fastqc.zip
│ ├── KPGP-00001_L3_R1_fastqc.html
│ ├── KPGP-00001_L3_R1_fastqc.zip
│ ├── KPGP-00001_L3_R2_fastqc.html
│ ├── KPGP-00001_L3_R2_fastqc.zip
│ ├── KPGP-00001_L4_R1_fastqc.html
│ ├── KPGP-00001_L4_R1_fastqc.zip
│ ├── KPGP-00001_L4_R2_fastqc.html
│ ├── KPGP-00001_L4_R2_fastqc.zip
│ ├── KPGP-00001_L5_R1_fastqc.html
│ ├── KPGP-00001_L5_R1_fastqc.zip
│ ├── KPGP-00001_L5_R2_fastqc.html
│ ├── KPGP-00001_L5_R2_fastqc.zip
│ ├── KPGP-00001_L6_R1_fastqc.html
│ ├── KPGP-00001_L6_R1_fastqc.zip
│ ├── KPGP-00001_L6_R2_fastqc.html
│ ├── KPGP-00001_L6_R2_fastqc.zip
│ ├── multiqc_data
│ │ ├── multiqc_data.json
│ │ ├── multiqc_fastqc.txt
│ │ ├── multiqc_general_stats.txt
│ │ ├── multiqc.log
│ │ └── multiqc_sources.txt
│ └── multiqc_report.html
└── script
├── bwa_index.sh
├── fastqc1.log
├── fastqc2.log
├── fastqc3.log
├── fastqc4.log
├── fastqc5.log
├── fastqc6.log
├── fastqc.sh
└── nohup.out
step2
cd /home/ubuntu/WGS_new/output
mkdir /home/ubuntu/WGS_new/output/bwa_mapping
cd /home/ubuntu/WGS_new/script
#比对的脚本如下
#!/bin/bash
bwa mem -t 4 -R '@RG\tID:KPGP-00001_L1\tPL:illumina\tSM:KPGP-00001' /home/ubuntu/WGS_new/input/genome/hg38 /home/ubuntu/WGS_new/input/KPGP-00001_L1_R1.fq.gz /home/ubuntu/WGS_new/input/KPGP-00001_L1_R2.fq.gz >/home/ubuntu/WGS_new/output/bwa_mapping/KPGP-00001_L1.sam && echo "KPGP-00001_L1 mapping is done"
bwa mem -t 4 -R '@RG\tID:KPGP-00001_L2\tPL:illumina\tSM:KPGP-00001' /home/ubuntu/WGS_new/input/genome/hg38 /home/ubuntu/WGS_new/input/KPGP-00001_L2_R1.fq.gz /home/ubuntu/WGS_new/input/KPGP-00001_L2_R2.fq.gz >/home/ubuntu/WGS_new/output/bwa_mapping/KPGP-00001_L2.sam && echo "KPGP-00001_L2 mapping is done"
bwa mem -t 4 -R '@RG\tID:KPGP-00001_L3\tPL:illumina\tSM:KPGP-00001' /home/ubuntu/WGS_new/input/genome/hg38 /home/ubuntu/WGS_new/input/KPGP-00001_L3_R1.fq.gz /home/ubuntu/WGS_new/input/KPGP-00001_L3_R2.fq.gz >/home/ubuntu/WGS_new/output/bwa_mapping/KPGP-00001_L3.sam && echo "KPGP-00001_L3 mapping is done"
bwa mem -t 4 -R '@RG\tID:KPGP-00001_L4\tPL:illumina\tSM:KPGP-00001' /home/ubuntu/WGS_new/input/genome/hg38 /home/ubuntu/WGS_new/input/KPGP-00001_L4_R1.fq.gz /home/ubuntu/WGS_new/input/KPGP-00001_L4_R2.fq.gz >/home/ubuntu/WGS_new/output/bwa_mapping/KPGP-00001_L4.sam && echo "KPGP-00001_L4 mapping is done"
bwa mem -t 4 -R '@RG\tID:KPGP-00001_L5\tPL:illumina\tSM:KPGP-00001' /home/ubuntu/WGS_new/input/genome/hg38 /home/ubuntu/WGS_new/input/KPGP-00001_L5_R1.fq.gz /home/ubuntu/WGS_new/input/KPGP-00001_L5_R2.fq.gz >/home/ubuntu/WGS_new/output/bwa_mapping/KPGP-00001_L5.sam && echo "KPGP-00001_L5 mapping is done"
bwa mem -t 4 -R '@RG\tID:KPGP-00001_L6\tPL:illumina\tSM:KPGP-00001' /home/ubuntu/WGS_new/input/genome/hg38 /home/ubuntu/WGS_new/input/KPGP-00001_L6_R1.fq.gz /home/ubuntu/WGS_new/input/KPGP-00001_L6_R2.fq.gz >/home/ubuntu/WGS_new/output/bwa_mapping/KPGP-00001_L6.sam && echo "KPGP-00001_L6 mapping is done"
#运行脚本
nohup bash bwa_mapping.sh &
比对后生成sam文件,将sam文件转换成二进制格式的bam文件
#转换的脚本如下
#!/bin/bash
for i in $(seq 1 6)
do
samtools view -Sb /home/ubuntu/WGS_new/output/bwa_mapping/KPGP-00001_L${i}.sam >/home/ubuntu/WGS_new/output/bwa_mapping/KPGP-00001_L${i}.bam && echo "KPGP-00001_L${i} samt2bam is done"
done
#运行脚本
nohup bash sam2bam.sh &
#运行结束后,再把sam格式文件删除,其实可以不删除,但是太占储存空间了
cd /home/ubuntu/WGS_new/output/bwa_mapping
rm *.sam
结果如下:
├── input
│ ├── genome
│ │ ├── hg38.amb
│ │ ├── hg38.ann
│ │ ├── hg38.bwt
│ │ ├── hg38.fa
│ │ ├── hg38.pac
│ │ ├── hg38.sa
│ │ └── nohup.out
│ ├── KPGP-00001_L1_R1.fq.gz
│ ├── KPGP-00001_L1_R2.fq.gz
│ ├── KPGP-00001_L2_R1.fq.gz
│ ├── KPGP-00001_L2_R2.fq.gz
│ ├── KPGP-00001_L3_R1.fq.gz
│ ├── KPGP-00001_L3_R2.fq.gz
│ ├── KPGP-00001_L4_R1.fq.gz
│ ├── KPGP-00001_L4_R2.fq.gz
│ ├── KPGP-00001_L5_R1.fq.gz
│ ├── KPGP-00001_L5_R2.fq.gz
│ ├── KPGP-00001_L6_R1.fq.gz
│ ├── KPGP-00001_L6_R2.fq.gz
│ └── nohup.out
├── output
│ ├── bwa_mapping
│ │ ├── KPGP-00001_L1.bam
│ │ ├── KPGP-00001_L2.bam
│ │ ├── KPGP-00001_L3.bam
│ │ ├── KPGP-00001_L4.bam
│ │ ├── KPGP-00001_L5.bam
│ │ └── KPGP-00001_L6.bam
│ └── fastqc_result
│ ├── KPGP-00001_L1_R1_fastqc.html
│ ├── KPGP-00001_L1_R1_fastqc.zip
│ ├── KPGP-00001_L2_R1_fastqc.html
│ ├── KPGP-00001_L2_R1_fastqc.zip
│ ├── KPGP-00001_L2_R2_fastqc.html
│ ├── KPGP-00001_L2_R2_fastqc.zip
│ ├── KPGP-00001_L3_R1_fastqc.html
│ ├── KPGP-00001_L3_R1_fastqc.zip
│ ├── KPGP-00001_L3_R2_fastqc.html
│ ├── KPGP-00001_L3_R2_fastqc.zip
│ ├── KPGP-00001_L4_R1_fastqc.html
│ ├── KPGP-00001_L4_R1_fastqc.zip
│ ├── KPGP-00001_L4_R2_fastqc.html
│ ├── KPGP-00001_L4_R2_fastqc.zip
│ ├── KPGP-00001_L5_R1_fastqc.html
│ ├── KPGP-00001_L5_R1_fastqc.zip
│ ├── KPGP-00001_L5_R2_fastqc.html
│ ├── KPGP-00001_L5_R2_fastqc.zip
│ ├── KPGP-00001_L6_R1_fastqc.html
│ ├── KPGP-00001_L6_R1_fastqc.zip
│ ├── KPGP-00001_L6_R2_fastqc.html
│ ├── KPGP-00001_L6_R2_fastqc.zip
│ ├── multiqc_data
│ │ ├── multiqc_data.json
│ │ ├── multiqc_fastqc.txt
│ │ ├── multiqc_general_stats.txt
│ │ ├── multiqc.log
│ │ └── multiqc_sources.txt
│ └── multiqc_report.html
└── script
├── bwa_index.sh
├── bwa_mapping.sh
├── fastqc1.log
├── fastqc2.log
├── fastqc3.log
├── fastqc4.log
├── fastqc5.log
├── fastqc6.log
├── fastqc.sh
├── nohup.out
└── sam2bam.sh
step3
对生成的bam文件按position排序
#脚本如下
#!/bin/bash
for i in $(seq 1 6)
do
samtools sort -@ 4 -m 4G -O bam -o /home/ubuntu/WGS_new/output/bwa_mapping/KPGP-00001_L${i}.sorted.bam /home/ubuntu/WGS_new/output/bwa_mapping/KPGP-00001_L${i}.bam && echo "/KPGP-00001_L${i}.sorted.bam is done"
done
此时所有结果如下
├── input
│ ├── genome
│ │ ├── hg38.amb
│?? │?? ├── hg38.ann
│?? │?? ├── hg38.bwt
│?? │?? ├── hg38.fa
│?? │?? ├── hg38.pac
│?? │?? ├── hg38.sa
│?? │?? └── nohup.out
│?? ├── KPGP-00001_L1_R1.fq.gz
│?? ├── KPGP-00001_L1_R2.fq.gz
│?? ├── KPGP-00001_L2_R1.fq.gz
│?? ├── KPGP-00001_L2_R2.fq.gz
│?? ├── KPGP-00001_L3_R1.fq.gz
│?? ├── KPGP-00001_L3_R2.fq.gz
│?? ├── KPGP-00001_L4_R1.fq.gz
│?? ├── KPGP-00001_L4_R2.fq.gz
│?? ├── KPGP-00001_L5_R1.fq.gz
│?? ├── KPGP-00001_L5_R2.fq.gz
│?? ├── KPGP-00001_L6_R1.fq.gz
│?? ├── KPGP-00001_L6_R2.fq.gz
│?? └── nohup.out
├── output
│?? ├── bwa_mapping
│?? │?? ├── index
│?? │?? ├── KPGP-00001_L1.bam
│?? │?? ├── KPGP-00001_L1.sorted.bam
│?? │?? ├── KPGP-00001_L2.bam
│?? │?? ├── KPGP-00001_L2.sorted.bam
│?? │?? ├── KPGP-00001_L3.bam
│?? │?? ├── KPGP-00001_L3.sorted.bam
│?? │?? ├── KPGP-00001_L4.bam
│?? │?? ├── KPGP-00001_L4.sorted.bam
│?? │?? ├── KPGP-00001_L5.bam
│?? │?? ├── KPGP-00001_L5.sorted.bam
│?? │?? ├── KPGP-00001_L6.bam
│?? │?? └── KPGP-00001_L6.sorted.bam
│?? └── fastqc_result
│?? ├── KPGP-00001_L1_R1_fastqc.html
│?? ├── KPGP-00001_L1_R1_fastqc.zip
│?? ├── KPGP-00001_L2_R1_fastqc.html
│?? ├── KPGP-00001_L2_R1_fastqc.zip
│?? ├── KPGP-00001_L2_R2_fastqc.html
│?? ├── KPGP-00001_L2_R2_fastqc.zip
│?? ├── KPGP-00001_L3_R1_fastqc.html
│?? ├── KPGP-00001_L3_R1_fastqc.zip
│?? ├── KPGP-00001_L3_R2_fastqc.html
│?? ├── KPGP-00001_L3_R2_fastqc.zip
│?? ├── KPGP-00001_L4_R1_fastqc.html
│?? ├── KPGP-00001_L4_R1_fastqc.zip
│?? ├── KPGP-00001_L4_R2_fastqc.html
│?? ├── KPGP-00001_L4_R2_fastqc.zip
│?? ├── KPGP-00001_L5_R1_fastqc.html
│?? ├── KPGP-00001_L5_R1_fastqc.zip
│?? ├── KPGP-00001_L5_R2_fastqc.html
│?? ├── KPGP-00001_L5_R2_fastqc.zip
│?? ├── KPGP-00001_L6_R1_fastqc.html
│?? ├── KPGP-00001_L6_R1_fastqc.zip
│?? ├── KPGP-00001_L6_R2_fastqc.html
│?? ├── KPGP-00001_L6_R2_fastqc.zip
│?? ├── multiqc_data
│?? │?? ├── multiqc_data.json
│?? │?? ├── multiqc_fastqc.txt
│?? │?? ├── multiqc_general_stats.txt
│?? │?? ├── multiqc.log
│?? │?? └── multiqc_sources.txt
│?? └── multiqc_report.html
└── script
├── bam_index.sh
├── bwa_index.sh
├── bwa_mapping.sh
├── fastqc1.log
├── fastqc2.log
├── fastqc3.log
├── fastqc4.log
├── fastqc5.log
├── fastqc6.log
├── fastqc.sh
├── nohup.out
└── sam2bam.sh
以上的步骤就完成了比对的过程
在此基础上,做一些以下的横向探索:
1.分别利用 samtools和 bedtools 对测序深度和覆盖度进行统计,并可视化
2.把bam文件按染色体分成小文件,并用IGV查看
3.bam格式转化为bw格式看测序深度分布
4.测序深度和GC含量的关系
1.分别利用 samtools和 bedtools 对测序深度和覆盖度进行统计,并可视化
用samtools 对测序深度进行统计
#!/bin/bash
for i in $(seq 1 6)
do
samtools depth /home/ubuntu/WGS_new/output/bwa_mapping/KPGP-00001_L${i}.sorted.bam 1>/home/ubuntu/WGS_new/output/bwa_mapping/samtools_depth/KPGP-00001_L${i}.sorted.bam.depth 2>&1 && echo "KPGP-00001_L${i}.sorted.bam depth count is done"
done
这个软件得到的文件格式如下
chr1 9999 4
chr1 10000 5
chr1 10001 51
chr1 10002 161
chr1 10003 250
chr1 10004 278
chr1 10005 365
chr1 10006 390
chr1 10007 391
chr1 10008 397
chr1 10009 406
chr1 10010 407
chr1 10011 414
chr1 10012 415
chr1 10013 417
chr1 10014 420
chr1 10015 420
chr1 10016 419
chr1 10017 421
chr1 10018 422
chr1 10019 425
chr1 10020 427
chr1 10021 430
chr1 10022 432
chr1 10023 435
chr1 10024 435
chr1 10025 437
chr1 10026 439
chr1 10027 439
chr1 10028 440
chr1 10029 440
chr1 10030 442
...
...
第一列是染色体号,第二列是对应的单个碱基位置,第三列是单个碱基的深度
需要进一步统计到每个染色体对应的测序深度
这一步的python脚本如下
import sys
args=sys.argv
filename=args[1]
SumDict={}
for line in open(filename):
lineL=line.strip().split("\t")
ID=lineL[0]
depth=int(lineL[2])
if ID not in SumDict:
SumDict[ID]=depth
else:
SumDict[ID]+=depth
for k,v in SumDict.items():
print(k,v,sep="\t")
-----------------------------
nohup time python3 sumDepth.py /home/ubuntu/WGS_new/output/bwa_mapping/samtools_depth/KPGP-00001_L1.sorted.bam.depth > /home/ubuntu/WGS_new/output/bwa_mapping/samtools_depth/KPGP-00001_L1.SumOfDepth.txt &
nohup time python3 sumDepth.py /home/ubuntu/WGS_new/output/bwa_mapping/samtools_depth/KPGP-00001_L2.sorted.bam.depth > /home/ubuntu/WGS_new/output/bwa_mapping/samtools_depth/KPGP-00001_L2.SumOfDepth.txt &
nohup time python3 sumDepth.py /home/ubuntu/WGS_new/output/bwa_mapping/samtools_depth/KPGP-00001_L3.sorted.bam.depth > /home/ubuntu/WGS_new/output/bwa_mapping/samtools_depth/KPGP-00001_L3.SumOfDepth.txt &
nohup time python3 sumDepth.py /home/ubuntu/WGS_new/output/bwa_mapping/samtools_depth/KPGP-00001_L4.sorted.bam.depth > /home/ubuntu/WGS_new/output/bwa_mapping/samtools_depth/KPGP-00001_L4.SumOfDepth.txt &
nohup time python3 sumDepth.py /home/ubuntu/WGS_new/output/bwa_mapping/samtools_depth/KPGP-00001_L5.sorted.bam.depth > /home/ubuntu/WGS_new/output/bwa_mapping/samtools_depth/KPGP-00001_L5.SumOfDepth.txt &
nohup time python3 sumDepth.py /home/ubuntu/WGS_new/output/bwa_mapping/samtools_depth/KPGP-00001_L6.sorted.bam.depth > /home/ubuntu/WGS_new/output/bwa_mapping/samtools_depth/KPGP-00001_L6.SumOfDepth.txt &
得到的统计结果还需要把带“_”部分除掉
#!/bin/bash
for i in $(seq 1 6)
do
grep -v "_" /home/ubuntu/WGS_new/output/bwa_mapping/samtools_depth/KPGP-00001_L${i}.SumOfDepth.txt >/home/ubuntu/WGS_new/output/bwa_mapping/samtools_depth/KPGP-00001_L${i}.SumOfDepth.final.txt
done
然后将所有的生成文件排序后合并,脚本如下
cd /home/ubuntu/WGS_new/output/bwa_mapping/samtools_depth
#脚本
--------
#!/bin/bash
for i in $(seq 1 6)
do
sed -i "s/chr//" KPGP-00001_L${i}.SumOfDepth.final.txt #首次去掉chr,方便排序
sort -k1,1n KPGP-00001_L${i}.SumOfDepth.final.txt > KPGP-00001_L${i}.SumOfDepth.sort.final.txt #对第一列按数值排序
sed -i 's/^/chr/' KPGP-00001_L${i}.SumOfDepth.sort.final.txt #再将chr添加回来
done
paste KPGP-00001_L*.SumOfDepth.sort.final.txt >KPGP-00001_all.SumOfDepth.sort.final.txt
awk '{sum=$2+$$4+$6+$8+$10+$12;print $1,sum;}' KPGP-00001_all.SumOfDepth.sort.final.txt >KPGP-00001.SumOfDepth.final_count.txt
---------
#运行脚本
nohup bash paste_sort.sh &
-----------
sed -i '1 i Chrom\tsum_of_depth' KPGP-00001.SumOfDepth.final_count.txt
cat KPGP-00001.SumOfDepth.final_count.txt
-----------
Chrom sum_of_depth
chrM 34591025
chrX 4093132439
chrY 125469175
chr1 6482954037
chr2 6376037101
chr3 5286606057
chr4 5330867456
chr5 4949134249
chr6 4420827669
chr7 4173524521
chr8 3842370943
chr9 3228529809
chr10 3626998764
chr11 3548159595
chr12 3490214776
chr13 2618593673
chr14 2359781279
chr15 2098133774
chr16 2341297030
chr17 1911455487
chr18 2082861061
chr19 1341657686
chr20 1741460772
chr21 1203547746
chr22 988334122
sed -i 's/ /\t/' KPGP-00001.SumOfDepth.final_count.txt
以上就将所有的染色体的覆盖位点的测序深度总和统计出来了
接下来计算覆盖度
因为samtools depth计算的是所有覆盖的每个碱基的深度,所以将对应染色体的统计信息提取,并计算有多少个碱基,得到的碱基数就是对应的覆盖长度。
所以有如下脚本统计
#!/bin/bash
for i in $(seq 1 22)
do
grep "chr${i} " /home/ubuntu/WGS_new/output/bwa_mapping/samtools_depth/KPGP-00001_L1.sorted.bam.depth |wc -l >/home/ubuntu/WGS_new/output/bwa_mapping/samtools_depth/chr${i}_cov.txt
sed -i "1 i chr${i}" /home/ubuntu/WGS_new/output/bwa_mapping/samtools_depth/chr${i}_cov.txt
done
grep "chrX " /home/ubuntu/WGS_new/output/bwa_mapping/samtools_depth/KPGP-00001_L1.sorted.bam.depth |wc -l >/home/ubuntu/WGS_new/output/bwa_mapping/samtools_depth/chrX_cov.txt #x染色体
sed -i "1 i chrX" /home/ubuntu/WGS_new/output/bwa_mapping/samtools_depth/chrX_cov.txt
grep "chrY " /home/ubuntu/WGS_new/output/bwa_mapping/samtools_depth/KPGP-00001_L1.sorted.bam.depth |wc -l >/home/ubuntu/WGS_new/output/bwa_mapping/samtools_depth/chrY_cov.txt #y染色体
sed -i "1 i chrY" /home/ubuntu/WGS_new/output/bwa_mapping/samtools_depth/chrY_cov.txt
grep "chrM " /home/ubuntu/WGS_new/output/bwa_mapping/samtools_depth/KPGP-00001_L1.sorted.bam.depth |wc -l >/home/ubuntu/WGS_new/output/bwa_mapping/samtools_depth/chrM_cov.txt #线粒体
sed -i "1 i chrM" /home/ubuntu/WGS_new/output/bwa_mapping/samtools_depth/chrM_cov.txt
这里计算的是一个lane的coverage,应该要计算所有6个lane的coverage,再求mean值,或者提前将6个lane的bam文件merge后再处理。此处由于系统存储空间问题,只对一个lane作简单统计。
sort -m chr*_cov.txt|sed -n 1~2p> chr_cov1.txt
sort -m chr*_cov.txt|sed -n 2~2p> chr_cov2.txt
paste chr_cov*.txt>chr_cov_all.txt
sed 's/chr//' chr_cov_all.txt|sort -k1,1n|sed 's/^/chr/'|sed '1 i Chrom\tcoverage' >chr_cov.txt
cat chr_cov.txt
Chrom coverage
chrM 16569
chrX 151545600
chrY 4004642
chr1 224300366
chr2 235466712
chr3 194391189
chr4 185704231
chr5 176555626
chr6 164731880
chr7 155128428
chr8 141501464
chr9 118343356
chr10 129732215
chr11 131193733
chr12 130177621
chr13 95749291
chr14 87847379
chr15 81867181
chr16 78578328
chr17 78271424
chr18 77398752
chr19 54075815
chr20 62148856
chr21 38164294
chr22 36926516
计算参考序列染色体的长度
这里首先需要将hg38中的重复的N序列去除
首先想到的是
grep -v "N" hg38.fa >hg38.cut.fa
这里有一个小问题,就是如果N藏在正常ATGC序列之中的话,就很容易去除掉正常的碱基
所以改成sed命令
sed 's/N//g' hg38.fa >hg38.sed.fa #把所有的N替换成空
sed '/^$/d' hg38.sed.fa >hg38.final.fa #去除空行
sed是将N替换成空,并不是将含N的行除去
虽然两个命令得到结果一样,但还是选择后者是正确的。
写了2个python脚本分别统计1.hg38原始文件对应各条染色体的长度
2.hg38去除N之后的各条染色体的长度
3.hg38原始文件的各条染色体的N含量
脚本如下
import sys
args=sys.argv
filename=args[1]
seq_id={}
for line in open(filename):
line=line.strip()
if line.startswith(">"):
seq_name = line
if seq_name not in seq_id:
seq_id[seq_name]=''
else:
seq_id[seq_name]+=line.strip()
for k,v in seq_id.items():
seq_length=len(v)
name=k[1:]
print(name,seq_length,sep="\t")
---------------------------------------------------
#运行脚本
--------------------
nohup python3 count_length.py hg38.fa > hg38.chr_length.txt &
nohup python3 count_length.py hg38.final.fa > hg38.cutN.chr_length.txt &
-----------------------------------
import sys
args=sys.argv
filename=args[1]
seq_id={}
for line in open(filename):
line=line.strip()
if line.startswith(">"):
seq_name = line
if seq_name not in seq_id:
seq_id[seq_name]=''
else:
seq_id[seq_name]+=line
for k,v in seq_id.items():
N_count=v.count("N")
name=k[1:]
print(k,N_count,sep="\t")
--------------------------
#运行脚本
nohup python3 count_N.py hg38.fa >hg38.chr.countN.txt &
这两个脚本本身没有问题,而且对于fasta格式读取,把ID 和序列先构建字典的方式也是之前总结出来的
但是!!!!
对于大型的文件,先构建字典,在读取字典的过程会花费太多的时间,所以这两个脚本运行了如下的时间,程序还没有跑完
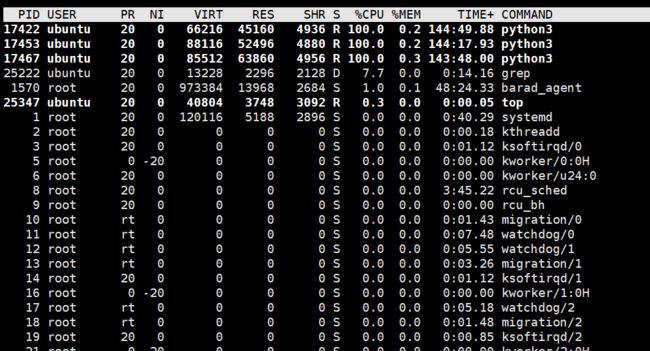
所以,需要修改代码,不要用这种构建字典的方式。
修改后的脚本如下:
#统计长度的脚本
import sys
args=sys.argv
filename=args[1]
seq_length=0
for line in open(filename):
line=line.strip()
if line.startswith(">"):
if seq_length:
print(seq_length)
seq_length=0
print(line)
else:
seq_length+=len(line)
print(seq_length)
--------------------------------------
#统计N数量的脚本
import sys
args=sys.argv
filename=args[1]
num_N=0
for line in open(filename):
line=line.strip()
if line.startswith(">"):
if num_N :
print (num_N)
num_N=0
print (line)
else:
num_N+=line.count("N")
print(num_N)
-----------
优化后的代码只花了2到3分钟就完成任务,可见代码优化的重要性。
因为得到结果不仅仅有chr1~22+x+y+M的信息,还有其他信息,所以需要提取
提取脚本如下
#!/bin/bash
for i in $(seq 1 22)
do
grep -A 1 ">chr${i}$" hg38.cutN.chr_length.txt > hg38.cutN.chr_length.final${i}.txt
grep -A 1 ">chr${i}$" count_N_new.txt > count_N_new.final${i}.txt
done
#!/bin/bash
for i in $(seq 1 22)
do
grep -A 1 ">chr${i}$" hg38.chr_length.txt >hg38.chr_length.final${i}.txt #提取>chr${i}$以及其下面一行,-A是after的意思,之所以是chr${i}+一个$是因为chr${i}后面加空行
才是想要的结果,如果不清楚可以cat -A 一下,再判断
grep -A 1 ">chr${i}$" hg38.cutN.chr_length.txt > hg38.cutN.chr_length.final${i}.txt
grep -A 1 ">chr${i}$" count_N_new.txt > count_N_new.final${i}.txt
done
grep -A 1 ">chrX$" hg38.chr_length.txt >hg38.chr_length.finalX.txt
grep -A 1 ">chrX$" hg38.cutN.chr_length.txt > hg38.cutN.chr_length.finalX.txt
grep -A 1 ">chrX$" count_N_new.txt > count_N_new.finalX.txt
grep -A 1 ">chrY$" hg38.chr_length.txt >hg38.chr_length.finalY.txt
grep -A 1 ">chrY$" hg38.cutN.chr_length.txt > hg38.cutN.chr_length.finalY.txt
grep -A 1 ">chrY$" count_N_new.txt > count_N_new.finalY.txt
grep -A 1 ">chrM$" hg38.chr_length.txt >hg38.chr_length.finalM.txt
grep -A 1 ">chrM$" hg38.cutN.chr_length.txt > hg38.cutN.chr_length.finalM.txt
grep -A 1 ">chrM$" count_N_new.txt > count_N_new.finalM.txt
rm hg38.chr_length.txt
rm hg38.cutN.chr_length.txt
rm count_N_new.txt
sort -m hg38.chr_length.final*.txt >hg38.chr_length.final_all.txt #将所有hg38.chr_length.final*.txt 文件合并,区别于paste,这个合并成列
sort -m hg38.cutN.chr_length.final*.txt >hg38.cutN.chr_length.final_all.txt
sort -m count_N_new.final*.txt >count_N_new.final_all.txt
sed -n 1~2p hg38.chr_length.final_all.txt >hg38.chr_length.final_all_1.txt #提取奇数行,1~2p中1是第一行,2p是从第一行开始,每2行输出,也就是输出奇数行
sed -n 2~2p hg38.chr_length.final_all.txt >hg38.chr_length.final_all_2.txt #提取偶数行,2~2p中1是第一行,2p是从第一行开始,每2行输出,也就是输出偶数行
paste hg38.chr_length.final_all_1.txt hg38.chr_length.final_all_2.txt > hg38.chr_length.final_all_3.txt #hg38每条染色体的长度,包括N
这里包括3个知识点:
- grep -A 1 string file 提取file中匹配到string的行和下面一行,1可以改成其他数字,表示多行
grep -B 1 string file 上面
grep -C 1 string file 上面和下面
2.cat -A 之后再找匹配的字符串比较准确
- sort -m 将文件合并成列,区别与paste
- sed -n 1~2p 提取奇数行
sed -n 2~2p 提取偶数行
当然awk也很容易实现
上面提取之后的文件排序,加列标题,然后合并
脚本如下:
sed 's/>chr//' count_N_new.final_all_3.txt |sort -k1,1n |sed 's/^/chr/'|sed '1 i Chrom\tN_Number' >N_number.txt #排序,然后添加列标题
sed 's/>chr//' hg38.cutN.chr_length.final_all_3.txt |sort -k1,1n |sed 's/^/chr/'|sed '1 i Chrom\tCut_N_length' >hg38.cutN.len.txt
sed 's/>chr//' hg38.chr_length.final_all_3.txt |sort -k1,1n |sed 's/^/chr/'|sed '1 i Chrom\tlength' >hg38.len.txt
paste hg38.len.txt hg38.cutN.len.txt N_number.txt |cut -f 1,2,4,6 >hg38.count_all_final.txt
less -S hg38.count_all_final.txt
-------------------------------------------------
Chrom length Cut_N_length N_Number
chrM 16569 16568 1
chrX 156040895 154893130 1147765
chrY 57227415 26415183 30812232
chr1 248956422 230481193 18475229
chr2 242193529 240548238 1645291
chr3 198295559 198100139 195420
chr4 190214555 189752667 461888
chr5 181538259 181265378 272881
chr6 170805979 170078724 727255
chr7 159345973 158970132 375841
chr8 145138636 144768136 370500
chr9 138394717 121790590 16604127
chr10 133797422 133263135 534287
chr11 135086622 134533742 552880
chr12 133275309 133138039 137270
chr13 114364328 97983128 16381200
chr14 107043718 90568149 16475569
chr15 101991189 84641348 17349841
chr16 90338345 81805944 8532401
chr17 83257441 82921074 336367
chr18 80373285 80089658 283627
chr19 58617616 58440758 176858
chr20 64444167 63944581 499586
chr21 46709983 40088683 6621300
chr22 50818468 39159777 11658691
--------------
结果一目了然
-------------------------------------------------------------------------------
最后再把测序深度、覆盖度、参考序列长度合并
cd /home/ubuntu/WGS_new/output/bwa_mapping/samtools_depth/new
ls -lh
------
total 12K
-rw-rw-r-- 1 ubuntu ubuntu 387 Aug 7 22:11 chr_cov.txt
-rw-rw-r-- 1 ubuntu ubuntu 834 Aug 7 21:51 hg38.count_all_final.txt
-rw-rw-r-- 1 ubuntu ubuntu 429 Aug 7 22:18 KPGP-00001.SumOfDepth.final_count.txt
-------
paste * |cut -f 1,2,4,5,6,8 >count_all.txt
cat count_all.txt
Chrom coverage length Cut_N_length N_Number sum_of_depth
chrM 16569 16569 16568 1 34591025
chrX 151545600 156040895 154893130 1147765 4093132439
chrY 4004642 57227415 26415183 30812232 125469175
chr1 224300366 248956422 230481193 18475229 6482954037
chr2 235466712 242193529 240548238 1645291 6376037101
chr3 194391189 198295559 198100139 195420 5286606057
chr4 185704231 190214555 189752667 461888 5330867456
chr5 176555626 181538259 181265378 272881 4949134249
chr6 164731880 170805979 170078724 727255 4420827669
chr7 155128428 159345973 158970132 375841 4173524521
chr8 141501464 145138636 144768136 370500 3842370943
chr9 118343356 138394717 121790590 16604127 3228529809
chr10 129732215 133797422 133263135 534287 3626998764
chr11 131193733 135086622 134533742 552880 3548159595
chr12 130177621 133275309 133138039 137270 3490214776
chr13 95749291 114364328 97983128 16381200 2618593673
chr14 87847379 107043718 90568149 16475569 2359781279
chr15 81867181 101991189 84641348 17349841 2098133774
chr16 78578328 90338345 81805944 8532401 2341297030
chr17 78271424 83257441 82921074 336367 1911455487
chr18 77398752 80373285 80089658 283627 2082861061
chr19 54075815 58617616 58440758 176858 1341657686
chr20 62148856 64444167 63944581 499586 1741460772
chr21 38164294 46709983 40088683 6621300 1203547746
chr22 36926516 50818468 39159777 11658691 988334122
awk 'BEGIN{OFS=FS="\t";}{if (NR>1){cov=$2/$4;print cov}}' count_all.txt >cov_percent.txt #计算覆盖率
awk 'BEGIN{OFS=FS="\t";}{if (NR>1){cov=$6/$2;print cov}}' count_all.txt >mean_depth.txt
sed -i '1 i cov_percent' cov_percent.txt
paste count_all.txt mean_depth.txt cov_percent.txt >count_all_done.txt
cat count_all_done.txt
Chrom coverage length Cut_N_length N_Number sum_of_depth mean_depth cov_percent
chrM 16569 16569 16568 1 34591025 2087.7 1.00006
chrX 151545600 156040895 154893130 1147765 4093132439 27.0092 0.978388
chrY 4004642 57227415 26415183 30812232 125469175 31.3309 0.151604
chr1 224300366 248956422 230481193 18475229 6482954037 28.903 0.973183
chr2 235466712 242193529 240548238 1645291 6376037101 27.0783 0.978875
chr3 194391189 198295559 198100139 195420 5286606057 27.1957 0.981277
chr4 185704231 190214555 189752667 461888 5330867456 28.7062 0.978665
chr5 176555626 181538259 181265378 272881 4949134249 28.0316 0.974017
chr6 164731880 170805979 170078724 727255 4420827669 26.8365 0.968563
chr7 155128428 159345973 158970132 375841 4173524521 26.9037 0.975834
chr8 141501464 145138636 144768136 370500 3842370943 27.1543 0.977435
chr9 118343356 138394717 121790590 16604127 3228529809 27.281 0.971695
chr10 129732215 133797422 133263135 534287 3626998764 27.9576 0.973504
chr11 131193733 135086622 134533742 552880 3548159595 27.0452 0.975173
chr12 130177621 133275309 133138039 137270 3490214776 26.8112 0.977764
chr13 95749291 114364328 97983128 16381200 2618593673 27.3484 0.977202
chr14 87847379 107043718 90568149 16475569 2359781279 26.8623 0.969959
chr15 81867181 101991189 84641348 17349841 2098133774 25.6285 0.967224
chr16 78578328 90338345 81805944 8532401 2341297030 29.7957 0.960545
chr17 78271424 83257441 82921074 336367 1911455487 24.4209 0.943927
chr18 77398752 80373285 80089658 283627 2082861061 26.9108 0.966401
chr19 54075815 58617616 58440758 176858 1341657686 24.8107 0.92531
chr20 62148856 64444167 63944581 499586 1741460772 28.0208 0.971917
chr21 38164294 46709983 40088683 6621300 1203547746 31.536 0.951997
chr22 36926516 50818468 39159777 11658691 988334122 26.7649 0.942971
知识补充:
1depth 测序深度
测序深度是指测序得到的总碱基数与待测基因组大小的比值,可以理解为基因组中每个碱基被测序到的平均次数。测序深度 = reads长度 × 比对的reads数目 / 参考序列长度。假设一个基因大小为2M,测序深度为10X,那么获得的总数据量为20M。
之所以测序深度=reads长度×比对的reads数目 / 参考序列长度,可以这么去理解,因为深度指的是每个碱基被测序到的平均次数。所以假设reads长度特别长,长到等于参考序列长度(当然这不可能)。那么,这时候reads数目就直接等于测序深度,但是reads长度远远没有参考序列长,所以才有了相当于reads长度/参考序列长度,这样的一个可以理解为矫正系数。
如果我们的研究目的仅仅是进行群体遗传分析等类型的分析。由于分析是通过计算基因组不同区域的多样性变化来推断进化选择压力,或构建进化树推断遗传分化关系,SNP检出率达到70~90%足以达到此类目的。所以,此类研究常见的测序深度选择在10X左右。
如果想检测个体的全基因组突变,来寻找某个特定功能突变,则我们推荐大于30X的测序深度,以保证接近于“毫无疏漏”。
如果测序样本是混样,例如,癌组织样本(癌细胞的细胞异质性很大),BSA分析中的群体混合池样本。那么在保证30X的测序深度的基础上,如果经费许可50X的测序深度更佳,以保证对混合池中低频变异的检测成功率(例如,新出现或即将消亡的癌细胞突变)。
2.coverage 覆盖度:
覆盖度是指测序获得的序列占整个基因组的比例。指的是基因组上至少被检测到1次的区域,占整个基因组的比例。当然,有些文章中也会将测序深度称为Coverage,容易给我们带来混淆。因此,我们还是需要根据语境来推断Coverage的意思。
我们假设基因组大小为G, 假定每次测序可从基因组任何位置上随机检测一个碱基。那么对于基因组上某一个固定碱基位置,在一次测序中,该位置被命中的概率为P (P=1/G)。我们将试验重复n次,相当于产生了n个碱基(n=c*G, c为测序深度)。 碱基的深度分布,相当于求该位置被测到0次,1次,…,n次的概率各是多少? 位点被检测到的n次的概率符合泊松分布。当然,由于概率极低,检测次数很大,所以这样的泊松分布接近于正态分布。
由于基因组中的高GC、重复序列等复杂结构的存在,测序最终拼接组装获得的序列往往无法覆盖有所的区域,这部分没有获得的区域就称为Gap。例如一个细菌基因组测序,覆盖度是98%,那么还有2%的序列区域是没有通过测序获得的。
上面统计所有染色体测序深度用的是python的脚本,这里也提供一个shell脚本,但是代码会相对长一点。
#!/bin/bash
for i in $(seq 1 22) #22对染色体
do
grep "chr${i} " /home/ubuntu/WGS_new/output/bwa_mapping/samtools_depth/KPGP-00001_L1.sorted.bam.depth |cut -f 3 |awk '{sum += $1;}END{print sum;}' >/home/ubuntu/WGS_new/output/bwa_mapping/samtools_depth/chr${i}.txt #注意END 的使用 |注意\t键的使用,脚本中可以直接键入tab,命令行下先Ctrl+V,再按tab键。这里之所以要加tab键,是因为,比如,如果>不加tab键,grep "chr1" 得到包括chr1,chr11,chr1_...>等。
sed -i "1 i chr${i}" /home/ubuntu/WGS_new/output/bwa_mapping/samtools_depth/chr${i}.txt # 1表示第一行,i表示插入,即在第一行前面插入新行,名称为chr${i}
done
grep "chrX " /home/ubuntu/WGS_new/output/bwa_mapping/samtools_depth/KPGP-00001_L1.sorted.bam.depth |cut -f 3 |awk '{sum += $1;}END{print sum;}' > /home/ubuntu/WGS_new/output/bwa_mapping/samtools_depth/chrx.txt #x染色体
grep "chrY " /home/ubuntu/WGS_new/output/bwa_mapping/samtools_depth/KPGP-00001_L1.sorted.bam.depth |cut -f 3 |awk '{sum += $1;}END{print sum;}' > /home/ubuntu/WGS_new/output/bwa_mapping/samtools_depth/chry.txt #y染色体
sed -i "1 i chrx" /home/ubuntu/WGS_new/output/bwa_mapping/samtools_depth/chrx.txt #添加列标题 chrx
sed -i "1 i chry" /home/ubuntu/WGS_new/output/bwa_mapping/samtools_depth/chry.txt
注意:awk命令要注意尽量冒号小写
awk '{sum += $1} END{print sum}' chr2.tmp
结果
3399
而
awk "{sum += $1} END{print sum}" chr2.tmp
报错
awk: cmd. line:1: {sum += } END{print sum}
awk: cmd. line:1: ^ syntax error
小总结
cut -f 可以提取特定列
cut -f 2 提取第2列
sed -n 2p 提取2行
sed -n 2,4p 提取2至3行
对于上诉得到的汇总结果存到count.txt中,并进行可视化,脚本如下
pdf()
library(ggplot2)
data <- read.table("count.txt",header = T)
#因为上一个命令,header=T已经将首行作为列标题,所以$1没法识别,用$加列名
data <-data[-1,] #将线粒体那行删除
data$Chrom <-factor(data$Chrom,levels = c(1:22,"X","Y")) #这一步很重要,这样横坐标就按顺序排列了
Chr <- data$Chrom
M <- data$mean_depth
C <- data$cov_percent
dt1 <- data.frame(Chr,M)
dt2 <- data.frame(Chr,C)
p1 <- ggplot(dt1,aes(x=Chr,y=M))+xlab("chrom ID")+ylab("mean_depth")+labs(title="KPGP-00001")+ylim(0,100)+geom_bar(aes(fill=Chr),stat = "identity")+theme_bw()+theme(plot.title = element_text(hjust = 0.5),panel.grid.major =element_blank(),panel.grid.minor = element_blank())
# 1 ggplot(dt1,aes(x=Chr,y=M,group=Chr))是构建一个画板
# 2 +xlab("chrom ID")+ylab("mean_depth") 是更改横纵坐标标签
# 3 +labs(title="KPGP-00001") 是增加主标题
# 4 ylim(0,100) 设置y轴刻度为1~100
# 5 +geom_bar(aes(fill=Chr),stat = "identity")是画出柱状图
# 6 theme_bw()为传统的白色背景和深灰色的网格线
# 7 theme是参数的设置 plot.title = element_text(hjust = 0.5) 是将title移到中间
# panel.grid.major =element_blank(),panel.grid.minor = element_blank()
#去除网格线
p2 <- ggplot(dt2,aes(x=Chr,y=C,group=1))+xlab("chrom ID")+ylab("coverage_percent")+labs(title="KPGP-00001")+ylim(0,1.5)+geom_line(aes(color=Chr))+geom_point(aes(color=Chr))+theme_bw()+theme(plot.title = element_text(hjust = 0.5),panel.grid.major =element_blank(),panel.grid.minor = element_blank())
p1
p2
dev.off()
最终结果如下
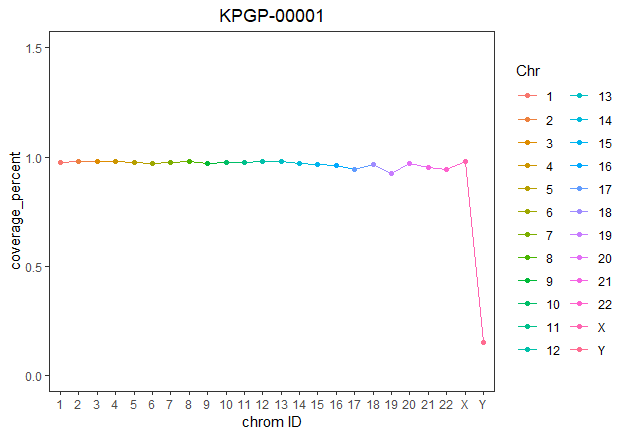
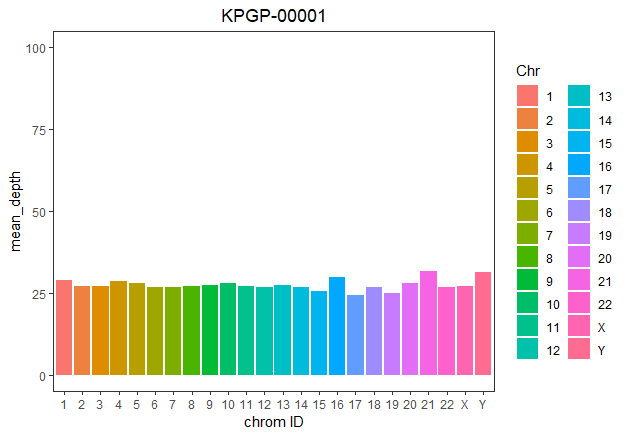
Y染色体的覆盖度特别低,不知道什么原因。
2.把bam文件按染色体分成小文件,并用IGV查看
bam文件太大了,有时候把bam文件拆分成小的文件是有必要的
这里用到一个工具bamtools,它的功能很多,这里选用split的功能
usage: bamtools [--help] COMMAND [ARGS]
Available bamtools commands:
convert Converts between BAM and a number of other formats
count Prints number of alignments in BAM file(s)
coverage Prints coverage statistics from the input BAM file
filter Filters BAM file(s) by user-specified criteria
header Prints BAM header information
index Generates index for BAM file
merge Merge multiple BAM files into single file
random Select random alignments from existing BAM file(s), intended more as a testing tool.
resolve Resolves paired-end reads (marking the IsProperPair flag as needed)
revert Removes duplicate marks and restores original base qualities
sort Sorts the BAM file according to some criteria
split Splits a BAM file on user-specified property, creating a new BAM output file for each value found
stats Prints some basic statistics from input BAM file(s)
See 'bamtools help COMMAND' for more information on a specific command.
这里只用lane1的bam文件做一个测试
nohup bamtools split -in KPGP-00001_L1.sorted.bam -reference &
#-reference指的是按reference拆分
拆分后,选取文件大小较小的chr19的bam文件,和chrY的文件(正好看看上一部布统计的覆盖度很低是不是统计错误)
#排序
samtools sort KPGP-00001_L1.sorted.REF_chrY.bam >chr_Y.sort.bam
samtools sort KPGP-00001_L1.sorted.REF_chr19.bam >chr_19.sort.bam
#建索引
samtools index chr_Y.sort.bam
samtools index chr_19.sort.bam
然后把文件导入IGV
先看一下chrY
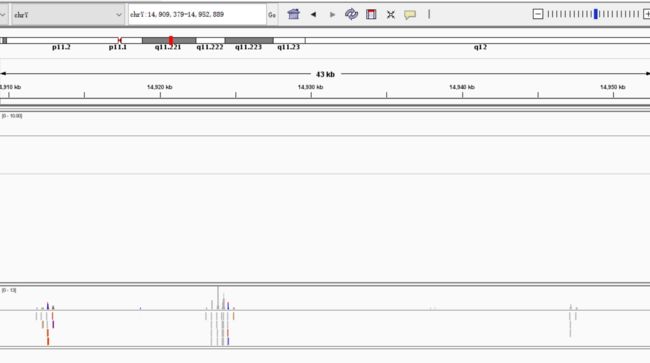
可见覆盖度确实很低,并不是之前统计的问题
再看看chr19
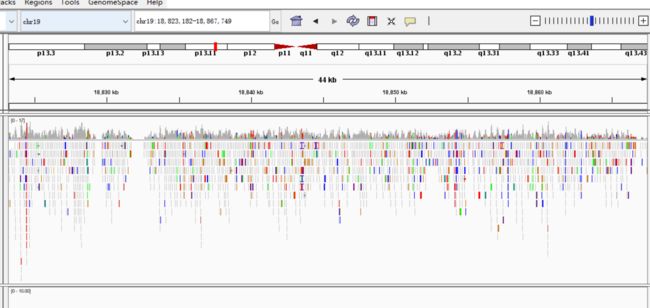
chr19的覆盖程度明显大很多
3.bam格式转化为bw格式看测序深度分布
bam文件很大,很多时候才看很不方便。有些时候,我们只需要我们的测序数据在全基因组的测序深度这一个值,并不需要具体某条reads的碱基序列,碱基质量值。这样只需要把bam文件压缩为bw格式。
同样是只用lane1做测试
这里用到的一个软件deeptools
用到它其中的一个工具bamCoverage
#首先还是要先建立索引
nohup samtools index KPGP-00001_L1.sorted.bam &
再就是转换格式
nohup bamCoverage -b KPGP-00001_L1.sorted.bam -o KPGP-00001_L1.sorted.bw &
bw格式的大小才200M,可以轻松的导入IGV,可以从整体看出测序的深度和覆盖度情况。
比如

很显然,y染色体覆盖度很小
4.测序深度和GC含量的关系
首先,把全基因组的bam文件用 mpileup模式输出,根据 1000bp 的窗口滑动来统计每个窗口的测到的碱基数,GC碱基数,测序总深度。因为涉及到测序深度,所以先把各个lane的bam文件merge,再拿merge后的bam文件得到结果的前面一百万行的数据做测试。
nohup samtools merge KPGP-00001.sorted.merge.bam KPGP-00001_L*.sorted.bam &
samtools mpileup -f /home/ubuntu/WGS_new/input/genome/new/hg38.fa KPGP-00001.sorted.merge.bam KPGP-00001_L*.sorted.bam |head -n 1000000 >1M_mpileup.txt
先看一下输出文件的大概格式(这个文件是之前用lane2的bam做的测试结果)

包括6列:
分别是:
染色体号、碱基位置、参考序列对应的碱基、比对上的reads数目(也就是depth)、比对的情况、比对的质量
命令中之所以加一个-f 参数就是为了提供参考序列fasta,使最终得到的结果文件的第3列是对应的碱基,如果不加,则变成了N,如下

既然加了参考序列,就序列提前对参考序列构建索引,软件要求的是faidx index。
回到正题
现在统计测序深度和GC含量的关系,理论上测序得到的reads 拼接的结果应该是和参考序列一致的,因此统计参考序列的GC含量实际上就是统计对应的测序本身的insert 片段的GC含量。然后与此同时统计测序的对应区间的测序深度的值,观察两者的关系。
python脚本如下
import sys
args=sys.argv
filename=args[1]
count_depth={}
count_base={}
count_number={}
count_all={}
with open (filename) as fh:
for line in fh:
lineL=line.strip().split("\t")
chr_ID=lineL[0]
base_position = int(lineL[1])
window_position = int(base_position/1000)
base=lineL[2]
base.upper() #大写
depth=int(lineL[3])
chr_win=chr_ID+" "+str(window_position)
if chr_win not in count_base :
count_base[chr_win]=0
# if chr_win not in count_depth: 因为两个字典是按同一个key,所以这条可以省略
count_depth[chr_win]=0
count_number[chr_win]=0
if base=="G" or base=="C":
count_base[chr_win]+=1
count_depth[chr_win]+=depth
count_number[chr_win]+=1
count_all[chr_win]=str(count_base[chr_win])+' '+str(count_depth[chr_win])+' '+str(count_number[chr_win]) #把3个字典的value合并成一个字符串作为一个新的字典的value
for k,v in count_all.items():
print(k,v,sep="\t")
这个脚本写的很长,我相信肯定改进的地方很大,但是这是我自己写的脚本,所以凑活用吧。
#总结一下:python字典计数的方式可大致分为两种写法
# 1. if key not in dict:
# dict[key]=1 或者其他初始值
# else:
# dict[key]+=1
# 2. if key not in dict:
# dict[key]='' 或者 0
# dict[key]+=1
#两种用法虽然没什么大的差别,但是有的情况下,2能用的起来,而1不行或更麻烦,比如此脚本情况
# 其实区别在于1中的if else,执行了if就不会执行else。而2中执行了if会继续执行下面的语句
python3 count.py 1M_mpileup.txt |sort -k2,2n >count_all.txt
head count_all.txt
---------------
chr1 9 0 3 1
chr1 10 0 425756 529
chr1 11 133 35354 952
chr1 12 600 46231 1000
chr1 13 575 41108 1000
chr1 14 582 51064 1000
chr1 15 537 30959 1000
chr1 16 541 28821 1000
chr1 17 621 51877 1000
chr1 18 537 34340 1000
-------------------------------------
第一列是染色体号,
第二列是对应的窗口第几个窗口(从10开始)
第三列是对应GC碱基数
第四列是总测序深度
第五列是每个窗口统计到的碱基总数
再分别计算一下GC%和mean depth就可以可视化了
在这之前还是先看看刚统计的结果,可以发现,以1000bp的窗口滑动,统计到的碱基数目并不为1000,有的甚至很低,其实这很容易理解。我的理解是,对于参考基因组,仍然是存在一些含N的重复序列,而这列序列在比对的过程中是不计算的,samtools mpileup 生成的文本中这部分就已经去除了,所以碱基的位置信息不是连续的。而脚本中1000bp是按碱基的位置信息除以1000取int来做的,所以统计到的碱基数目不一致是正常的。
接下来给出可视化的R脚本:
library(ggplot2)
dt <- read.table("count.txt") #读入数据
#head(dt)
dt$GC <- dt$V3/dt$V5 #得到GC含量
dt$depth <-dt$V4/dt$V5 #得到平均的depth
dt <- dt[dt$GC>0,] #把GC等于0的部分去除
dt <- dt[dt$depth<100,] #把depth大于100的去除
lm_eqn <- function(x,y){
m <- lm(y ~ x);
eq <- substitute(italic(y) == a + b %.% italic(x)*","~~italic(r)^2~"="~r2,
list(a = format(coef(m)[1], digits = 2),
b = format(coef(m)[2], digits = 2),
r2 = format(summary(m)$r.squared, digits = 3)))
as.character(as.expression(eq));
} #添加线性回归方程
p <- ggplot(dt, aes(x = GC, y = depth)) +xlim(c(0,0.8))+ylim(c(0,100))+
geom_smooth(method = "lm", se=FALSE, color="black", formula = y ~ x)+
geom_point()+geom_text(x=0.6,y=80,label = lm_eqn(dt$GC , dt$depth), parse = TRUE)+theme_bw()+theme(panel.grid = element_blank())
#作图
结果图如下
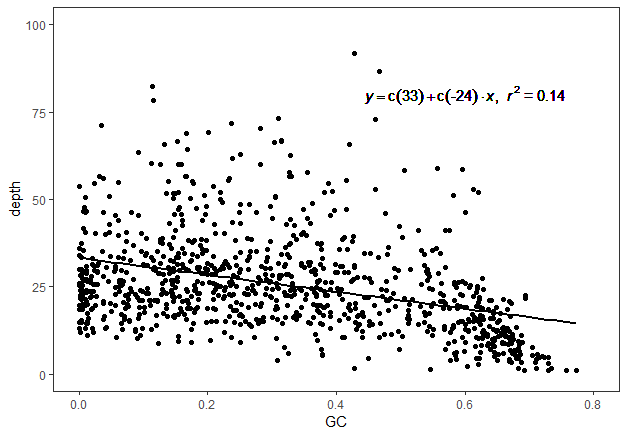
从这个结果看似乎是负相关,所以再取其他百万行的结果重复一遍,看看是否得到同样的结果
这是第2000000至3000000之间的图
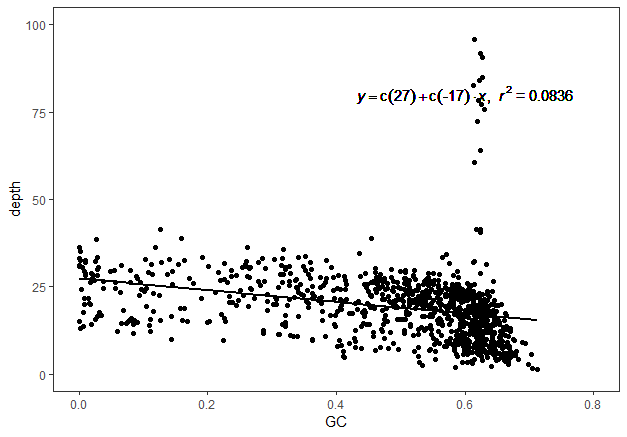
负相关,不知道什么原因,先不去管它
Part4 去重复(或标记重复序列)
以上对比对的横向探索结束
接下来继续纵向走,下一步是
去重复(或标记重复序列)
这里用两种软件:samtools和picard分别处理
1.samtools
这里本来应该是用samtools的markdup来标记或去除PCR重复,本来脚本已经写好开始运行了,但是报错了
脚本如下
#!/bin/bash
dir1=~/WGS_new/output/bwa_mapping
dir2=~/WGS_new/output/bwa_mapping/Dup
for i in $(seq 1 6)
do
samtools markdup -r $dir1/KPGP-00001_L${i}.sorted.bam $dir2/KPGP-00001_L${i}.sorted.rm.bam && echo "rm KPGP-00001_L${i} done"
samtools markdup -S $dir1/KPGP-00001_L${i}.sorted.bam $dir2/KPGP-00001_L${i}.sorted.mk.bam && echo "mk KPGP-00001_L${i} done"
done
报错信息如下
[markdup] error: no ms score tag.
[markdup] error: no ms score tag.
[markdup] error: no ms score tag.
[markdup] error: no ms score tag.
[markdup] error: no ms score tag.
[markdup] error: no ms score tag.
[markdup] error: no ms score tag.
仔细看了一下说明书
Usage: samtools markdup
Option:
-r Remove duplicate reads
-l INT Max read length (default 300 bases)
-S Mark supplemenary alignments of duplicates as duplicates (slower).
-s Report stats.
-T PREFIX Write temporary files to PREFIX.samtools.nnnn.nnnn.tmp.
-t Mark primary duplicates with the name of the original in a 'do' tag. Mainly for information and debugging.
--input-fmt-option OPT[=VAL]
Specify a single input file format option in the form
of OPTION or OPTION=VALUE
-O, --output-fmt FORMAT[,OPT[=VAL]]...
Specify output format (SAM, BAM, CRAM)
--output-fmt-option OPT[=VAL]
Specify a single output file format option in the form
of OPTION or OPTION=VALUE
--reference FILE
Reference sequence FASTA FILE [null]
-@, --threads INT
Number of additional threads to use [0]
The input file must be coordinate sorted and must have gone through fixmates with the mate scoring option on.
最后一行说到,必须gone through fixmates with the mate scoring option on
所以需要先fixmate
samtools fixmate KPGP-00001_L1.sorted.bam KPGP-00001_L1.sorted.fix.bam
然而又报错了
[bam_mating_core] ERROR: Coordinate sorted, require grouped/sorted by queryname
bam文件需要按quername排序,而samtools默认是按position排序的,而原始的bam文件为了减少储存,已经被我删除了。。。。
所以这一步没办法做了,因而samtools markdup去重就不做了,直接用picard的吧
2.picard
这里用GATK4的中的picard
#!/bin/bash
dir1=~/WGS_new/output/bwa_mapping
dir2=~/WGS_new/output/bwa_mapping/GATK/Dup
for i in $(seq 1 6)
do
#将重复序列删除,注意--REMOVE_DUPLICATES=true
gatk MarkDuplicates --REMOVE_DUPLICATES=true -I $dir1/KPGP-00001_L${i}.sorted.bam -M $dir2/KPGP-00001_L${i}.rm_metrics.txt -O $dir2/KPGP-00001_L${i}.rm.bam && echo "rm KPGP-00001_L${i} done"
#只是将重复标记,可以让后续软件识别,但是并没有删除
gatk MarkDuplicates -I $dir1/KPGP-00001_L${i}.sorted.bam -M $dir2/KPGP-00001_L${i}.mk_metrics.txt -O $dir2/KPGP-00001_L${i}.mk.bam && echo "mk KPGP-00001_L${i} done"
done
这里先解释为什么要去除重复,这部分节选自解螺旋的矿工公众号
首先,我们需要先理解什么是重复序列,它是如何产生的,以及为什么需要去除掉?要回答这几个问题,我们需要再次理解在建库和测序时到底发生了什么。
我们已经知道,在NGS测序之前都需要先构建测序文库:通过物理(超声)打断或者化学试剂(酶切)切断原始的DNA序列,然后选择特定长度范围的序列去进行PCR扩增并上机测序。
因此,这里重复序列的来源实际上就是由PCR过程中所引入的。因为所谓的PCR扩增就是把原来的一段DNA序列复制多次。可是为什么需要PCR扩增呢?如果没有扩增不就可以省略这一步了吗?
情况确实如此,但是很多时候我们构建测序文库时能用的细胞量并不会非常充足,而且在打断的步骤中也会引起部分DNA的降解,这两点会使整体或者局部的DNA浓度过低,这时如果直接从这个溶液中取样去测序就很可能漏掉原本基因组上的一些DNA片段,导致测序不全。而PCR扩增的作用就是为了把这些微弱的DNA多复制几倍乃至几十倍,以便增大它们在溶液中分布的密度,使得能够在取样时被获取到。所以这里大家需要记住一个重点,PCR扩增原本的目的是为了增大微弱DNA序列片段的密度,但由于整个反应都在一个试管中进行,因此其他一些密度并不低的DNA片段也会被同步放大,那么这时在取样去上机测序的时候,这些DNA片段就很可能会被重复取到相同的几条去进行测序。看到这里,你或许会觉得,那没必要去除不也应该可以吗?因为即便扩增了多几次,不也同样还是原来的那一段DNA吗?直接用来分析对结果也不会有影响啊!难道不是吗?
会有影响,而且有时影响会很大!最直接的后果就是同时增大了变异检测结果的假阴和假阳率。主要有几个原因:
DNA在打断的那一步会发生一些损失,主要表现是会引发一些碱基发生颠换变换(嘌呤-变嘧啶或者嘧啶变嘌呤),带来假的变异。PCR过程会扩大这个信号,导致最后的检测结果中混入了假的结果;
PCR反应过程中也会带来新的碱基错误。发生在前几轮的PCR扩增发生的错误会在后续的PCR过程中扩大,同样带来假的变异;
对于真实的变异,PCR反应可能会对包含某一个碱基的DNA模版扩增更加剧烈(这个现象称为PCR Bias)。因此,如果反应体系是对含有reference allele的模板扩增偏向强烈,那么变异碱基的信息会变小,从而会导致假阴。
PCR对真实的变异检测和个体的基因型判断都有不好的影响。GATK、Samtools、Platpus等这种利用贝叶斯原理的变异检测算法都是认为所用的序列数据都不是重复序列(即将它们和其他序列一视同仁地进行变异的判断,所以带来误导),因此必须要进行标记(去除)或者使用PCR-Free的测序方案(这种方案目前正变得越来越流行,特别是对于RNA-Seq来说尤为重要,现在著名的基因组学研究所——Broad Institute,基本都是使用PCR-Free的测序方案)。
跑完上面的流程的结果如下:
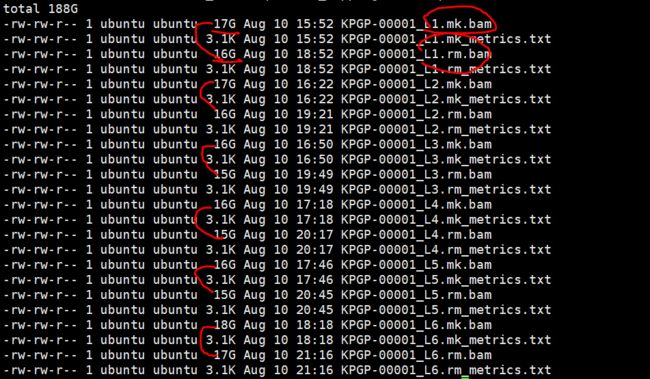
可以看出只是标记重复的(mk)文件是要比去除重复(rm)文件要大的。
再看一下之前的bam 文件
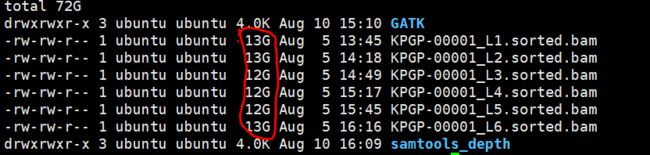
很显然这两个处理后的文件都是要比原始文件要大的。
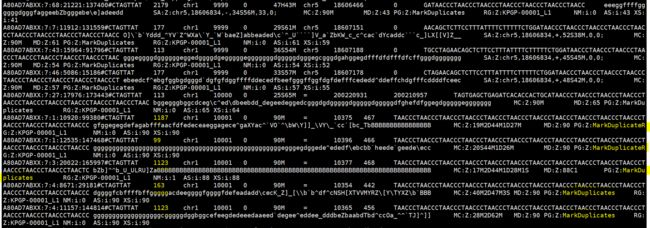
分别看看mk和rm生成的文件有什么不同
samtools view KPGP-00001_L1.mk.bam |head
结果如下
samtools view KPGP-00001_L1.rm.bam |head
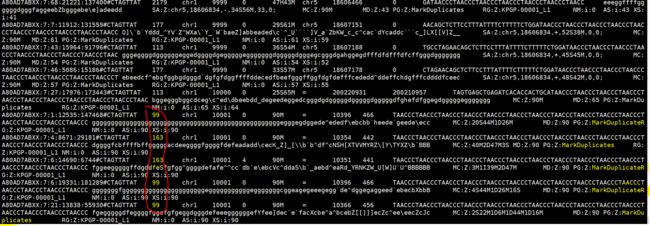
samtools view KPGP-00001_L1.sorted.bam |head
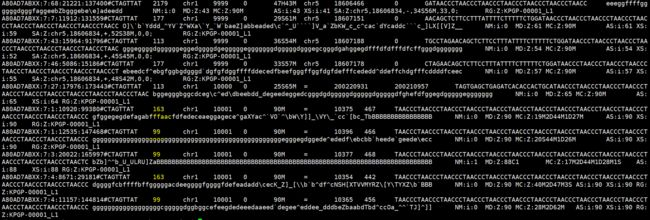
很明显,相比原始bam文件,rm多了一些markduplicate的字符,所以文件变大了
而,相比rm文件,mk,在flag值一列变大了,所以mk比rm文件大,这些通过文件内容很容易看出来
从下一步开始就直接用mk文件
因为如果 MarkDuplicates 这步把重复去掉,则会对 mate 信息产生影响,可以考虑在这步之后加上 FixMateInformation。
但是如果只是 MarkDuplicates而没有去除重复,则不会对mate信息产生影响,理论上不需要 FixMateInformation这一步
这个可以在哪得到论证呢
我们可以看看上面对3个bam文件查看的内容,对比flag值
(荧光标出来的部分)
mk.bam rm.bam sort.bam
1187 99 163
99 163 99
1123 163 99
163 99 163
1123 99 99
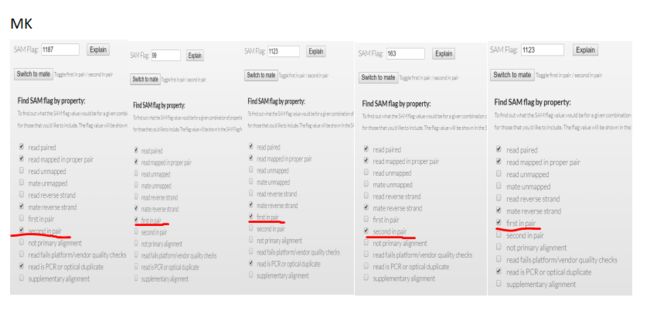
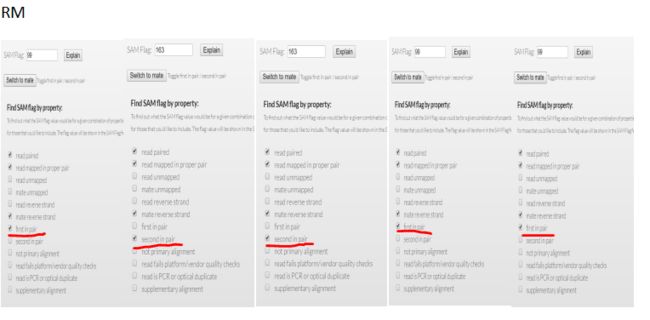
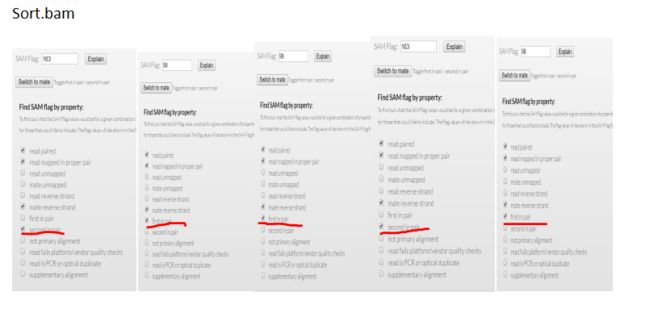
结果显而易见
Part5 碱基质量重校正
先下载一些必备数据库
#!/bin/bash
wget ftp://[email protected]/bundle/hg38/1000G_phase1.snps.high_confidence.hg38.vcf.gz
wget ftp://[email protected]/bundle/hg38/1000G_phase1.snps.high_confidence.hg38.vcf.gz.tbi
wget ftp://[email protected]/bundle/hg38/Mills_and_1000G_gold_standard.indels.hg38.vcf.gz
wget ftp://[email protected]/bundle/hg38/Mills_and_1000G_gold_standard.indels.hg38.vcf.gz.tbi
wget ftp://[email protected]/bundle/hg38/hapmap_3.3.hg38.vcf.gz
wget ftp://[email protected]/bundle/hg38/hapmap_3.3.hg38.vcf.gz.tbi
wget ftp://[email protected]/bundle/hg38/dbsnp_146.hg38.vcf.gz
wget ftp://[email protected]/bundle/hg38/dbsnp_146.hg38.vcf.gz.tbi
结果如下
total 5.1G
-rw-rw-r-- 1 ubuntu ubuntu 1.8G Aug 11 00:52 1000G_phase1.snps.high_confidence.hg38.vcf.gz
-rw-rw-r-- 1 ubuntu ubuntu 2.1M Aug 11 01:07 1000G_phase1.snps.high_confidence.hg38.vcf.gz.tbi
-rw-rw-r-- 1 ubuntu ubuntu 3.2G Aug 11 03:04 dbsnp_146.hg38.vcf.gz
-rw-rw-r-- 1 ubuntu ubuntu 2.4M Aug 11 03:20 dbsnp_146.hg38.vcf.gz.tbi
-rw-rw-r-- 1 ubuntu ubuntu 837 Aug 10 23:17 download.sh
-rw-rw-r-- 1 ubuntu ubuntu 60M Aug 11 01:08 hapmap_3.3.hg38.vcf.gz
-rw-rw-r-- 1 ubuntu ubuntu 1.5M Aug 11 01:08 hapmap_3.3.hg38.vcf.gz.tbi
-rw-rw-r-- 1 ubuntu ubuntu 20M Aug 11 01:07 Mills_and_1000G_gold_standard.indels.hg38.vcf.gz
-rw-rw-r-- 1 ubuntu ubuntu 1.5M Aug 11 01:07 Mills_and_1000G_gold_standard.indels.hg38.vcf.gz.tbi
在此之前先把之前下载的hg38.fa构建dict,注意dict和之前的bwa构建的index不一样
cd /home/ubuntu/WGS_new/input/genome/new
nohup gatk CreateSequenceDictionary -R hg38.fa -O hg38.dict &
创建了字典之后就可以继续了
#!/bin/bash
snp=/home/ubuntu/WGS_new/output/bwa_mapping/GATK/resource/dbsnp_146.hg38.vcf.gz
indel=/home/ubuntu/WGS_new/output/bwa_mapping/GATK/resource/Mills_and_1000G_gold_standard.indels.hg38.vcf.gz
#dir1=~/WGS_new/output/bwa_mapping
dir2=~/WGS_new/output/bwa_mapping/GATK/Dup
#dir3=/home/ubuntu/WGS_new/output/bwa_mapping/GATK/index
dir4=/home/ubuntu/WGS_new/output/bwa_mapping/GATK/resource
dir5=/home/ubuntu/WGS_new/output/bwa_mapping/GATK/recal
ref=~/WGS_new/input/genome/new/hg38.fa
for i in $(seq 1 6)
do
gatk BaseRecalibrator -R $ref -I $dir2/KPGP-00001_L${i}.mk.bam --known-sites $snp --known-sites $indel -O $dir5/KPGP-00001_L${i}.mk_recal.table
done
得到的table文件只是一个中间文件,还需要一步才能得到BQSR后的bam文件。
#!/bin/bash
snp=/home/ubuntu/WGS_new/output/bwa_mapping/GATK/resource/dbsnp_146.hg38.vcf.gz
indel=/home/ubuntu/WGS_new/output/bwa_mapping/GATK/resource/Mills_and_1000G_gold_standard.indels.hg38.vcf.gz
#dir1=~/WGS_new/output/bwa_mapping
dir2=~/WGS_new/output/bwa_mapping/GATK/Dup
#dir3=/home/ubuntu/WGS_new/output/bwa_mapping/GATK/index
dir4=/home/ubuntu/WGS_new/output/bwa_mapping/GATK/resource
dir5=/home/ubuntu/WGS_new/output/bwa_mapping/GATK/recal
ref=~/WGS_new/input/genome/new/hg38.fa
#for i in $(seq 1 6)
#do
#gatk BaseRecalibrator -R $ref -I $dir2/KPGP-00001_L${i}.mk.bam --known-sites $snp --known-sites $indel -O $dir5/KPGP-00001_L${i}.mk_recal.table
#done
for i in $(seq 1 6)
do
gatk ApplyBQSR -R $ref -I $dir2/KPGP-00001_L${i}.mk.bam -bqsr $dir5/KPGP-00001_L${i}.mk_recal.table -O $dir5/KPGP-00001_L${i}.mk_bqsr.bam
done
好了,到了这一步,利用生成的高质量的bam,接下来就是用4种工具来找变异,分别是GATK、bcftools、freebayes、varscan
再这之前先将各个lane生成的bam文件合并
nohup samtools merge KPGP-00001_bqsr.merge.bam *.bam
Part6 找变异
1.GATK
先对上一步生成的merge的bam文件建立索引
nohup samtools index KPGP-00001_bqsr.merge.bam &
然后是使用GATK的HaplotypeCaller
#!/bin/bash
snp=/home/ubuntu/WGS_new/output/bwa_mapping/GATK/resource/dbsnp_146.hg38.vcf.gz
indel=/home/ubuntu/WGS_new/output/bwa_mapping/GATK/resource/Mills_and_1000G_gold_standard.indels.hg38.vcf.gz
#dir1=~/WGS_new/output/bwa_mapping
dir2=~/WGS_new/output/bwa_mapping/GATK/Dup
#dir3=/home/ubuntu/WGS_new/output/bwa_mapping/GATK/index
dir4=/home/ubuntu/WGS_new/output/bwa_mapping/GATK/resource
dir5=/home/ubuntu/WGS_new/output/bwa_mapping/GATK/recal
dir6=/home/ubuntu/WGS_new/output/bwa_mapping/GATK/vcf
ref=~/WGS_new/input/genome/new/hg38.fa
gatk HaplotypeCaller -R $ref -I $dir5/KPGP-00001_bqsr.merge.bam --dbsnp $snp -O $dir6/KPGP-00001_bqsr.merge.vcf
2.bcftools
#!/bin/bash
snp=/home/ubuntu/WGS_new/output/bwa_mapping/GATK/resource/dbsnp_146.hg38.vcf.gz
indel=/home/ubuntu/WGS_new/output/bwa_mapping/GATK/resource/Mills_and_1000G_gold_standard.indels.hg38.vcf.gz
#dir1=~/WGS_new/output/bwa_mapping
dir2=~/WGS_new/output/bwa_mapping/GATK/Dup
#dir3=/home/ubuntu/WGS_new/output/bwa_mapping/GATK/index
dir4=/home/ubuntu/WGS_new/output/bwa_mapping/GATK/resource
dir5=/home/ubuntu/WGS_new/output/bwa_mapping/GATK/recal
dir6=/home/ubuntu/WGS_new/output/bwa_mapping/GATK/vcf
ref=~/WGS_new/input/genome/new/hg38.fa
samtools mpileup -ugf $ref $dir5/KPGP-00001_bqsr.merge.bam | bcftools call --thread 6 -v -m -O z -o $dir6/KPGP-00001_bqsr.vcf.gz
最终用两个软件得到的vcf大小如下:

用GATK Ht得到的vcf大小为988M
用bcftools得到的大小为663M
Part7 简单的可视化比对文件和变异的结果
因为比对的bam文件太大了,导入IGV并不可取
所以可以简化一个就只看某个基因的变异情况
比如BRCA1
1.首先从gtf文件中得到BRCA1基因的坐标
从genecode下载的基因组的注释文件gtf
#注意gz文件不能直接grep
zcat gencode.v28.annotation.gtf.gz |grep "BRCA1"|grep -v "transcript_id"
#结果如下
chr17 HAVANA gene 43044295 43170245 . - . gene_id "ENSG00000012048.21"; gene_type "protein_coding"; gene_name "BRCA1"; level 2; tag "overlapping_locus"; havana_gene "OTTHUMG00000157426.14";
所以得到BRCA1的基因坐标为
chr17:43044295-43170245
2.从总的bam文件中提取BRCA1的bam文件
用samtools view来提取BRCA1的bam文件
samtools view -h KPGP-00001_bqsr.merge.bam chr17:43044295-43170245 |samtools sort -o BRCA1.bam -
#提取,再排序
再对生成的bam文件建立索引(IGV必须的步骤)
samtools index BRCA1.bam
3.提取BRCA1区间的vcf文件
grep "chr17 " KPGP-00001_bqsr.vcf |awk '{if ($2>43044295&&$2<43170245) {print $0;} }' >BRCA1_bcftools.vcf
grep "chr17 " KPGP-00001_bqsr.merge.vcf |awk '{if ($2>43044295&&$2<43170245) {print $0;} }' >BRCA1_gatk.vcf
可以先了解一下这2个vcf有什么不同
wc -l BRCA1_gatk.vcf
#282 BRCA1_gatk.vcf
wc -l BRCA1_bcftools.vcf
#271 BRCA1_bcftools.vcf
似乎是gatk比bcftools找的变异有一些不同
先找到不同变异的位置
python脚本如下
import sys
args=sys.argv
filename1=args[1] #基准
filename2=args[2]
Alist=[]
Blist=[]
for line in open (filename1):
lineL=line.strip().split("\t")
Alist.append(lineL[1]) #把bcftools中的position存到Alist中,注意不能用“+”这种添加方式
for line in open (filename2):
lineL=line.strip().split("\t")
pos=lineL[1]
if pos not in Alist:
Blist.append(pos)
for i in Blist:
print(i)
找出gatk找到的,而bcf中没有的
python3 dis.py BRCA1_bcftools.vcf BRCA1_gatk.vcf
43045922
43046757
43050618
43054744
43067255
43077746
43079788
43098030
43098525
43102948
43106118
43111100
43112722
43112723
43112726
43112732
43112733
43112734
43112736
43112738
43114390
43118761
43123323
43128817
43133637
43142964
43149269
43155699
43166140
python3 dis.py BRCA1_bcftools.vcf BRCA1_gatk.vcf |wc -l
29
找出bcftools找到的,而GATK中没有的
python3 dis.py BRCA1_gatk.vcf BRCA1_bcftools.vcf
43050613
43050615
43052817
43067836
43077762
43086105
43089550
43089552
43090048
43100595
43100596
43102357
43104058
43104083
43123322
43142960
43142966
43166150
python3 dis.py BRCA1_gatk.vcf BRCA1_bcftools.vcf |wc -l
18
可见GATK和bcftools分别除了这29个和18个,剩下的253个变异的位点找到的是一致的
这些不同并不是最重要的,因为我们还没有对变异质控过滤
看几个共同的
bcftools的vcf

GATK的vcf
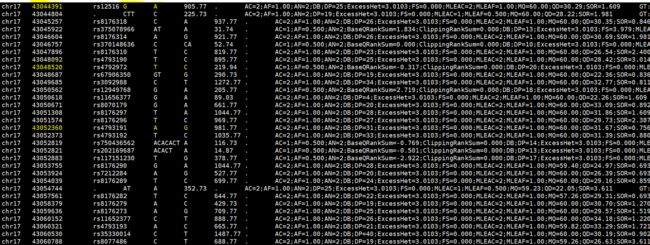
4.将BRCA1区间的bam文件导入IGV
43044391

43048520
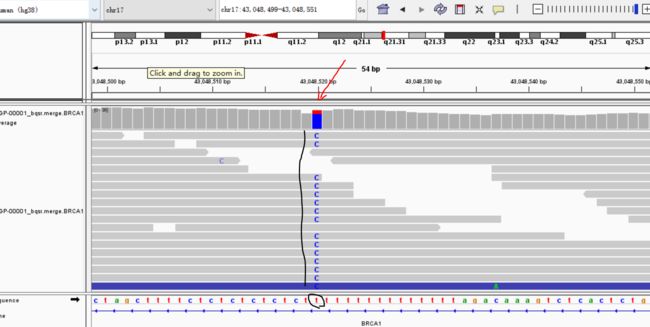
43052360
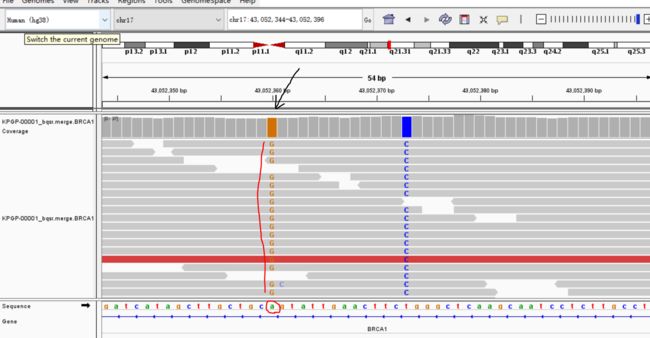
以上就是对比对结果和变异结果的简单可视化
Part8 比对以及变异结果的质控
1.raw和clean的fastq文件都需要使用fastqc质控。
这个做过了
2.比对的各个阶段的bam文件都可以质控。
比对各个的阶段的bam文件都是可以质控的
这里因为我只还有bqsr后的merge的bam文件,所以就拿这个来做个测试
使用的工具是samtools的flagstat
nohup samtools flagstat KPGP-00001_bqsr.merge.bam >KPGP-00001_bqsr.merge.stat &
结果如下:
1077675587 + 0 in total (QC-passed reads + QC-failed reads)
0 + 0 secondary
1460481 + 0 supplementary
88369576 + 0 duplicates
1073568441 + 0 mapped (99.62% : N/A)
1076215106 + 0 paired in sequencing
538107553 + 0 read1
538107553 + 0 read2
1047425796 + 0 properly paired (97.32% : N/A)
1068909862 + 0 with itself and mate mapped
3198098 + 0 singletons (0.30% : N/A)
12264590 + 0 with mate mapped to a different chr
6236962 + 0 with mate mapped to a different chr (mapQ>=5)
简单解读一下:
1077675587 是总的reads数
1460481 是supplementary(但这里指的是总的reads中不是pair-end的那部分reads数目)
88369576 是重复(虽然这个bam是做过去重步骤的,但是重复序列只是标记,没有remove,所以这里有是正常的)
1073568441 是指map上的总reads数,可见匹配率99.62%
1076215106 是指pair-end的reads数,基本上都是,总reads数减掉这个就是supplementary
538107553 + 0 read1
538107553 + 0 read2 双端测序两端的reads数目
1047425796指的是完美匹配的reads数目,完美匹配率也还是有97.32%
1068909862 指的是两端都匹配上的reads数目
3198098 指只有一端匹配上的reads数目
12264590 两端都匹配到不同的区域的reads
6236962 同上一个,只是其中比对质量>=5的reads的数量
3.使用qualimap对wes的比对bam文件总结测序深度及覆盖度。
关于对bam文件总结测序深度及覆盖度,前面已经自己专门写了脚本做了这件事
但是是有软件可以做这件事的,比如qualimap,这里就不重复了
4.变异结果的质控
质控的目的是指通过一定的标准,最大可能性的剔除假阳性的结果,并尽可能的保留最多的数据
对变异结果质控一般可以分为两种
第一种:软过滤
也就是使用GATK的VQSR,它使用机器学习的方法,利用多个不同数据的特征训练一个模型(高斯混合模型)对变异的结果进行质控
但是软过滤是有一定的条件的:
(1)需要一个精心准备的已知变异集合,作为训练质控模型的真集,比如:Hapmap,OMINI,1000G,dbsnp等这些国际性项目的数据
(2)要求新检测的结果中有足够多的变异,不然VQSR在进行模型训练时,会因为可用的变异位点数目不足而无法进行。
很多非人的物种在完成变异检测后,没法使用VQSR进行质控。同样的,一些小panel、外显子测序,由于最后的变异位点数目不够而无法进行VQSR。
全基因组分析或者多个样本的全外显子组分析可以使用此法。
第二种:硬过滤
简单的说就是对一些过滤指标一定的阈值,然后一刀切
主要涉及一下的一些过滤指标:
QualByDepth(QD):QD是变异质量值除以覆盖深度得到的比值
FisherStrand(FS):FS是一种通过Fisher检验得到的pValue转换而来的值,它描述的是在测序或者比对的时候,对只含有变异的read以及只含有参考序列碱基的read是否有明显的链特异性(strand bias)
StrandOddRatio (SOR):是对连特异性的一种描述
RMSMappingQuality(MQ):MQ这个值是所有比对到该位点上的read的比对质量值的均方根
MappingQualityRankSumTest(MQRankSum)
ReadPosRankSumTest (ReadPosRankSum)
其实这六个指标也就是VQSR进行质控模型训练的指标,它可以在一定的程度上描述每个变异的质量,因此,使用这些指标来进行硬过滤在一定程度上是合理的。
现在的问题是:什么样的指标是合理的。
常用的指标是:
snp:
QD<2 || MQ<40 || FS>60 || SOR >3.0 ||MQRankSum <-12.5|| ReadPosRankSum<-8.0
Indel:
QD<2 || FS>200.0 || SOR >10.0 ||MQRankSum <-12.5|| ReadPosRankSum<-8.0
至于为什么选择这个参数,**碱基矿工**在《GATK4.0和全基因组数据分析实践(下)》(https://mp.weixin.qq.com/s?__biz=MzAxOTUxOTM0Nw==&mid=2649798455&idx=1&sn=67a7407980a57ce138948eb46992b603&scene=21#wechat_redirect)已经说的很清楚了
其实简单的说就是用一些国际大项目的样本,查看样本VQSR前后的这些指标的分布,来确定阈值
这里给出我用GATK VQSR的脚本,我使用的是GATK4.0的版本
#!/bin/bash
#dir1=~/WGS_new/output/bwa_mapping
dir2=~/WGS_new/output/bwa_mapping/GATK/Dup
#dir3=/home/ubuntu/WGS_new/output/bwa_mapping/GATK/index
dir4=/home/ubuntu/WGS_new/output/bwa_mapping/GATK/resource
dir5=/home/ubuntu/WGS_new/output/bwa_mapping/GATK/recal
dir6=/home/ubuntu/WGS_new/output/bwa_mapping/GATK/vcf
GATK_bundle=~/WGS_new/output/bwa_mapping/GATK/resource
ref=~/WGS_new/input/genome/new/hg38.fa
gatk VariantRecalibrator -R $ref -V $dir6/KPGP-00001_bqsr.merge.vcf\
-resource hapmap,known=false,training=true,truth=true,prior=15.0:$GATK_bundle/hapmap_3.3.hg38.vcf.gz\
-resource 1000G,known=false,training=true,truth=false,prior=10.0:$GATK_bundle/1000G_phase1.snps.high_confidence.hg38.vcf.gz\
-resource dbsnp,known=true,traning=false,truth=false,prior=6.0:$GATK_bundle/dbsnp_146.hg38.vcf.gz\
-an DP -an QD -an FS -an SOR -an ReadPosRankSum -an MQRankSum\
-mode SNP\
-tranches-file $dir6/KPGP-00001.GATK.snps.trache\
-tranche 100.0 -tranche 99.9 -tranche 99.0 -tranche 95.0 -tranche 90.0\
-rscript-file $dir6/KPGP-00001.GATK.snps.plots.R\
-O $dir6/KPGP-00001.GATK.snps.recal
gatk ApplyVQSR \
-R $ref \
-V $dir6/KPGP-00001_bqsr.merge.vcf \
-ts-filter-level 99.0 \
-tranches-file $dir6/KPGP-00001.GATK.snps.trache\
-recal-file $dir6/KPGP-00001.GATK.snps.recal\
-mode SNP \
-O $dir6/KPGP-00001.GATK.VQSR.snps.vcf.gz
因为INDEL mode没有跑成功,并不清楚原因,先不管他,后面就用SNP注释
关于硬过滤,这里给出用法
#SNP mode
#选出SNP
GATK SelectVariants -select-type SNP -v *.raw.vcf -O *.snp.vcf
#对SNP 过滤
GATK VariantFiltration -V *.snp.vcf --filter-expression "QD<2 || MQ<40 || FS>60 || SOR >3.0 ||MQRankSum <-12.5|| ReadPosRankSum<-8.0" --filter-name "PASS" -O *.snp.filter.vcf
#INDEL mode
#选出INDEL
GATK SelectVariants -select-type INDEL -v *.raw.vcf -O *.indel.vcf
#对INDEL过滤
GATK VariantFiltration -V *.indel.vcf --filter-expression "QD<2 || FS>200.0 || SOR >10.0 ||MQRankSum <-12.5|| ReadPosRankSum<-8.0" --filter-name "PASS" -O *.indel.filter.vcf
再看看VQSR的结果
grep -v "#" KPGP-00001.GATK.VQSR.snps.vcf|wc -l
4565613
---------------------
grep "PASS" KPGP-00001.GATK.VQSR.snps.vcf > KPGP-00001.GATK.VQSR.pass.snps.vcf
wc -l KPGP-00001.GATK.VQSR.pass.snps.vcf
3252051 KPGP-00001.GATK.VQSR.pass.snps.vcf
可见,过滤了大约100多万的变异
Part9 变异的结果的注释
常用的注释软件有VEP、snpEFF、annovar
annovar注释
annovar是个perl编写的命令行工具,需要edu邮箱才能注册
annovar下载的时候会自带一些数据库存放在humandb文件夹中,但主要是hg19版本的,所以一般需要自己更新下载一些数据库
- 3种变异的注释方式
(1)Gene-based Annotation(基于基因的注释):基于基因的注释解释了变异与已知基因的直接关系,以及对其产生的功能性的影响
(2)Region-based Annotation(基于区域的注释):基于区域的注释看起来更像是一个区域的查询(这个区域可以是一个单一的位点),在一个数据库中,它不在乎位置的精确匹配,不在乎核苷酸的识别。基于区域的注释,揭示变异与不同基因组特定段的关系。例如:它是否落在已知的保守基因组区域。
(3)Filter-based Annotation(基于过滤的注释):则给出这个变异的一系列信息,如: population frequency in different populations 和various types of variant-deleteriousness prediction scores,这些可被用来过滤掉一些公共的probably
nondeleterious variants。filter-based和region-based的区别在于,filter-based针对mutation(核苷酸的变化),region-based针对染色体上的位置。如在全基因组数据中的变异频率,可以用1000g2015aug、kaviar_20150923等数据库;在全外显子组数据中的变异频率,可使用exac03、esp6500siv2_all等;在孤立或低代表人群中的变异频率可使用ajews等数据库。
2.对应注释方式的数据库下载
这3种注释方式即可以分别进行,也可以同时进行,无论哪种方式,对应方式的数据库都是需要下载的。
(1)gene-based annotation:
refGene
annotate_variation.pl -downdb -buildver hg38 -webfrom annovar refGene humandb/
(2)region-based annotation:
保守基因组元件注释:
phastConsElements46way
annotate_variation.pl -downdb -buildver hg38 -webfrom annovar phastConsElements46way humandb/
转录因子结合位点注释:
tfbsConsSites
annotate_variation.pl -downdb -buildver hg38 -webfrom annovar tfbsConsSites humandb/
细胞遗传学区段(cytogenetic band):
cytoBand
annotate_variation.pl -downdb -buildver hg38 -webfrom annovar cytoBand humandb/
Identify variants disrupting microRNAs and snoRNAs:
wgRna
annotate_variation.pl -downdb -buildver hg38 -webfrom annovar wgRna humandb/
.
.
.
.
.
等等,具体更多的可以查看Annovar documentation
(3)filter-based annotation:
由于这一类注释方式的数据库实在太多了,Annovar官方给了总结如下:
For frequency of variants in whole-genome data:
1000g2015aug: latest 1000 Genomes Project dataset with allele frequencies in six populations including ALL, AFR (African), AMR (Admixed American), EAS (East Asian), EUR (European), SAS (South Asian). These are whole-genome
variants.
kaviar_20150923: latest Kaviar database with 170 million variants from 13K genomes and 64K exomes.
hrcr1: latest Haplotype Reference Consortium database with 40 million variants from 32K samples in haplotype reference consortium
cg69: allele frequency in 69 human subjects sequenced by Complete Genomics. useful to exclude platform specific variants.
gnomad_genome: allele frequency in gnomAD database whole-genome sequence data on multiple populations.
For frequency of variants in whole-exome data:
exac03: latest Exome Aggregation Consortium dataste with allele frequencies in ALL, AFR (African), AMR (Admixed American), EAS (East Asian), FIN (Finnish), NFE (Non-finnish European), OTH (other), SAS (South Asian).
esp6500siv2: latest NHLBI-ESP project with 6500 exomes. Three separate key words are used for 3 population groupings: esp6500siv2_all, esp6500siv2_ea, esp6500siv2_aa.
gnomad_exome: allele frequency in gnomAD database whole-exome sequence data on multiple populations.
For frequency of variants in isolated or less represented populations:
ajews: common alleles in ashkenazi jews
TMC-SNPDB: common alleles in Indian populations
gme: GME (Greater Middle East Variome) allele frequency, including ALL, NWA (northwest Africa), NEA (northeast Africa), AP (Arabian peninsula), Israel, SD (Syrian desert), TP (Turkish peninsula) and CA (Central Asia).
???: I strongly encourage ANNOVAR users to donate your own allelel frequency database on isolated/special populations to be shared to worldwide users
For functional prediction of variants in whole-genome data:
gerp++: functional prediction scores for 9 billion mutations based on selective constraints across human genome. You can optionally use gerp++gt2 instead since it includes only RS score greater than 2, which provides high sensitivity while still strongly enriching for truly constrained sites
cadd: Combined Annotation Dependent Depletion score for 9 billion mutations. It is basically constructed by a support vector machine trained to differentiate 14.7 million high-frequency human-derived alleles from 14.7 million simulated variants, using ~70 different features. For known indels, use caddindel.
cadd13: CADD version 1.3.
dann: functional prediction score generated by deep learning, using the identical set of training data as cadd but with much improved performance than cadd.
fathmm: a hidden markov model to predict the functional importance of both coding and non-coding variants (that is, two separate scores are provided) on 9 billion mutations.
eigen: a spectral approach integrating functional genomic annotations for coding and noncoding variants on 9 billion mutations, without labelled training data (that is, unsupervised approach)
gwava: genome-wide annotation of variants that supports prioritization of noncoding variants by integrating various genomic and epigenomic annotations on 9 billion mutations.
For functional prediction of variants in whole-exome data:
dbnsfp30a: this dataset already includes SIFT, PolyPhen2 HDIV, PolyPhen2 HVAR, LRT, MutationTaster, MutationAssessor, FATHMM, MetaSVM, MetaLR, VEST, CADD, GERP++, DANN, fitCons, PhyloP and SiPhy scores, but ONLY on coding variants
For functional prediction of splice variants:
dbscsnv11: dbscSNV version 1.1 for splice site prediction by AdaBoost and Random Forest, which score how likely that the variant may affect splicing
spidex: deep learning based prediction of splice variants. Unlike dbscsnv11, these variants could be far away from canonical splice sites
For disease-specific variants:
clinvar_20160302: ClinVar database with separate columns (CLINSIG CLNDBN CLNACC CLNDSDB CLNDSDBID) for each variant (Please check the download page for the latest version, or read below for creating your own most updated version)
cosmic70: the latest COSMIC database with somatic mutations from cancer and the frequency of occurence in each subtype of cancer. For more updated cosmic, see instructions below on how to make them.
icgc21: International Cancer Genome Consortium version 21 mutations.
nci60: NCI-60 human tumor cell line panel exome sequencing allele frequency data
For variant identifiers:
snp142: dbSNP version 142
avsnp142: an abbreviated version of dbSNP 142 with left-normalization by
ANNOVAR developers. (Please check the download page for the latest version)
3.分别进行3种注释方式的代码和同时进行3种注释方式的代码
分别的:
以gene-based annotation为代表:
convert2annovar.pl -format vcf4old ~/WGS_new/output/bwa_mapping/GATK/vcf/KPGP-00001.GATK.VQSR.pass.snps.vcf >~/WGS_new/output/bwa_mapping/GATK/vcf/KPGP-00001.GATK.VQSR.pass.snps.annovar
annotate_variation.pl -buildver hg38 --outfile ~/WGS_new/output/bwa_mapping/GATK/vcf/KPGP-00001.GATK.VQSR.pass.snps.anno ~/WGS_new/output/bwa_mapping/GATK/vcf/KPGP-00001.GATK.VQSR.pass.snps.annovar humandb
其他的可以参考annovar的说明书:
#download annotation databases from ANNOVAR or UCSC and save to humandb/ directory
annotate_variation.pl -downdb -webfrom annovar refGene humandb/
annotate_variation.pl -buildver mm9 -downdb refGene mousedb/
annotate_variation.pl -downdb -webfrom annovar esp6500siv2_all humandb/
#gene-based annotation of variants in the varlist file (by default --geneanno is ON)
annotate_variation.pl -buildver hg19 ex1.avinput humandb/
#region-based annotate variants
annotate_variation.pl -regionanno -buildver hg19 -dbtype cytoBand ex1.avinput humandb/
annotate_variation.pl -regionanno -buildver hg19 -dbtype gff3 -gff3dbfile tfbs.gff3 ex1.avinput humandb/
#filter rare or unreported variants (in 1000G/dbSNP) or predicted deleterious variants
annotate_variation.pl -filter -dbtype 1000g2015aug_all -maf 0.01 ex1.avinput humandb/
annotate_variation.pl -filter -buildver hg19 -dbtype snp138 ex1.avinput humandb/
annotate_variation.pl -filter -dbtype dbnsfp30a -otherinfo ex1.avinput humandb/
得到的结果如下:
首先看一下全基因组范围的:
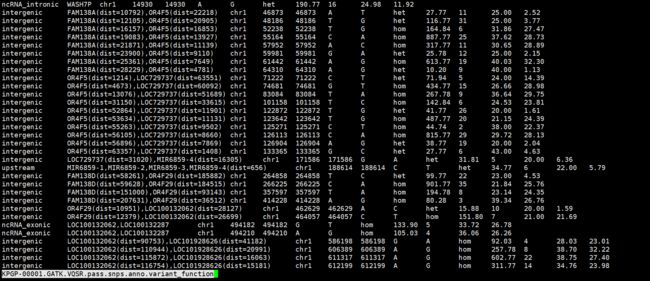
第一列包含的值有:
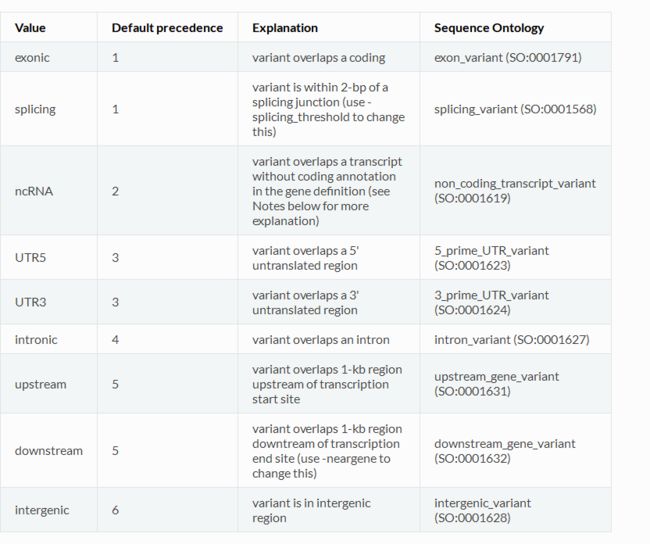
再重点看一下外显子区域的:
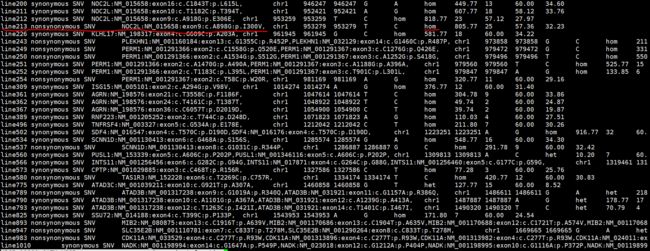
简单解释一下红色线画出的部分:
这是一个非同义突变(DNA 片 段中有时某个碱基对的突变改变所编码 氨基酸的改变)的SNV,发生在NOC2L这个基因上,gene ID 是NM_015658,这个变异发生在该基因的第9个exon上面,A变成G,蛋白方面是氨基酸I变成V,这是一个纯和的变异
同时进行的:
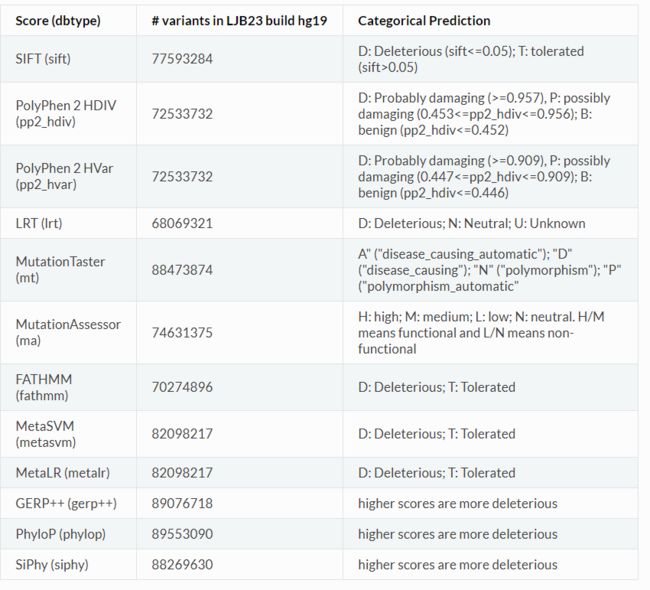
nohup table_annovar.pl ~/WGS_new/output/bwa_mapping/GATK/vcf/KPGP-00001.GATK.VQSR.pass.snps.annovar ~/soft/annovar/humandb/ -buildver hg38 -out anpanno -remove -protocol refGene,cytoBand,dbnsfp30a,EAS.sites.2015_08,avsnp142 -operation g,r,f,f,f -nastring . -csvout &
在这里,filter-based annotation选择了3个数据库,分别是
dbnsfp30a
1000g2015aug(EAS.sites.2015_08)
avsnp142
其中avsnp142是dbSNP数据库的简单版
1000g2015aug(EAS.sites.2015_08):千人基因组计划,这里使用的EAS,也就是东亚人群的数据库,因为我是在韩国个人基因组计划中下载的数据,所以使用的是东亚的数据库。
dbnsfp30a:这个数据库包括了IFT, PolyPhen2 HDIV, PolyPhen2 HVAR, LRT, MutationTaster, MutationAssessor, FATHMM, MetaSVM, MetaLR, VEST, CADD, GERP++, DANN, fitCons, PhyloP and SiPhy scores,通过评分,用于判断变异是不是有害的变异。这些分值只针对编码区的基因。
所以接下来要做的就是先过滤掉千人基因组计划和dbsnp中含有的变异(这并不合理,但是突变实在太多,只是作为观察的对象,或者说这部分是我感兴趣的)
然后用dbnsfp来判断变异是否有害
得到的结果中居然没有千人基因组计划数据库中变异,所以还是按dbsnp数据库进行过滤
由于全基因组的变异实在太多,这里就只把外显子区域的变异提取出来单独看,并且只保留非同义突变
grep '"exonic"' anpanno.hg38_multianno.csv |grep '"nonsynonymous SNV"' >anpanno.hg38_multianno.exon.nonsynonymous.csv
然后是过滤掉dbsnp中已经有的突变
import sys
args=sys.argv
filename=args[1]
with open (filename) as fh:
for line in fh:
if line.startswith("C"):
print(line)
lineL=line.strip().split(",")
if lineL[-1]==".":
print(line)
python3 ex.py anpanno.hg38_multianno.exon.nonsynonymous.new.csv >exon.nonsynonymous.filter.csv
wc -l exon.nonsynonymous.filter.csv
472 exon.nonsynonymous.filter.csv
这样最终得到472个变异
将文件导入R,并筛选
data <- read.csv("exon.nonsynonymous.filter.csv")
data <- data[data$SIFT_pred=="D",]
data <- data[data$Polyphen2_HDIV_pred=="D",]
data <- data[data$PROVEAN_pred=="D",]
data <- data[data$LRT_pred=="D",]
data <- data[data$fathmm.MKL_coding_pred=="D",]
data <- data[data$MetaLR_pred=="D",]
data <- data[data$MetaSVM_pred=="D",]
data <- data[data$FATHMM_pred=="D",]
data <- data[data$Polyphen2_HVAR_pred=="D",]
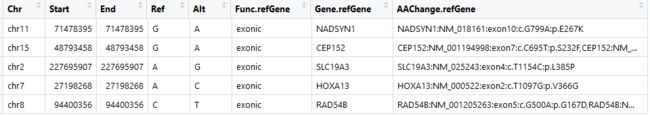
data <-data[,c("Chr","Start","End","Ref","Alt","Func.refGene","Gene.refGene","AAChange.refGene")]
最终得到这些指标一致认为有害的变异有5个
到此,整个WGS的流程的简单的学习算暂时结束,下游分析其实还有太多更复杂的地方,所以需要学习的地方还有很多很多
慢慢努力,一步一个脚印