本文接着每日文献:2018-02-27,上文探讨方法,本文是具体代码
为了解基因组存在T-DNA插入时,即基因组构成为AC而样本基因组为ABC的情况得到的测序结果在序列比对的时候的可能情况,因此需要先要使用模拟数据进行探索。
第一步:构建参考序列和实际序列。这一部分会用到samtools,emboss和entrez-direct, 都可以通过conda安装
用efecth下载参考基因组
mkdir -p refs
efetch -db=nuccore -format=fasta -id=AF086833 | seqret --filter -sid AF086833 > refs/AF086833.
fa
从参考基因组提取其中部分序列用作参考序列,而下载的参考基因组则被当成实际的基因组
# 提取1~5000, 8000~
cat refs/AF086833.fa | seqret -filter -sbegin 1 -send 5000 > part1.fa
cat refs/AF086833.fa | seqret -filter -sbegin 8000 > part2.fa
# 合并
cat part1.fa part2.fa| union -filter > refs/ref.fa
第二步:模拟测序结果。这一步用到dwgsim,也可以用conda安装
mkdir data
dwgsim -e 0.02 -E 0.02 -d 350 -1 100 -2 100 -s 50 -r 0 -R 0 -N 10000 -c 0 refs/AF086833.fa data/data
解释dwgsim的参数,-e和-E为测序仪的系统错误率, -d表示文库大小, -1和-2表示短读长度(这里就是文库大小350bp,PE100), 而-s则表示文库大小的波动情况,-r和-R表示基因组的突变率, -N表示输出的短读数, -c表示输出数据类型(0为illumina, 1为SOLiD,2为Ion Torrent)。最后会在data文件下生成以data为前缀的几个文件。
第三步:回贴到参考序列。所用工具为bwa和samtools
# 建立索引
bwa index refs/ref.fa
# 比对
mkdir align
bwa mem refs/ref.fa data/data.bwa.read1.fastq.gz data/data.bwa.read2.fastq.gz| samtools sort > align/data.bwa.bam
samtools index align/data.bwa.bam
第四步:使用IGV和samtools探索比对结果. samtools是处理SAM/BAM格式的常用工具,而IGV则是可视化利器。首先用samtools的flagstat统计比对的总体情况:
$ samtools flagstat align/data.bwa.bam
20000 + 0 in total (QC-passed reads + QC-failed reads)
0 + 0 secondary
0 + 0 supplementary
0 + 0 duplicates
15906 + 0 mapped (79.53% : N/A)
20000 + 0 paired in sequencing
10000 + 0 read1
10000 + 0 read2
15662 + 0 properly paired (78.31% : N/A)
15662 + 0 with itself and mate mapped
244 + 0 singletons (1.22% : N/A)
0 + 0 with mate mapped to a different chr
0 + 0 with mate mapped to a different chr (mapQ>=5)
不难大部分序列(~80%)都是正确成对(properly paired),其中properly paired的解释为"0x2 PROPER_PAIR .. each segment properly aligned according to the aligner",也就是两个序列都能在基因组上找到自己的位置,最常见的两类flags就是"83,163"和"99和147",也就是和参考序列反向互补。

那么余下的20%序列是什么情况?我们可以通过管道的方式进行简单的统计
$ samtools view -F 0x2 align/data.bwa.bam | cut -f 2 | sort | uniq -c | sort -k1,1nr
1925 141
1925 77
65 137
65 69
62 121
62 181
59 117
59 185
58 133
58 73
其中大部分序列是77和141,也就是说两条reads都没有比对到参考基因组上, 也就是SAM格式中的第3,6,7列为"*",第4,5,8,9列表示为"0"

剩下的"69,137","117,185"和"73,133","121,181"表示两条reads中只有一条,即flags为137,185和73,121的reads能比对到参考基因组上。

如果统计这些单边比对reads的位置信息,就会发现他们的位置是在4651~5214, 也就缩小搜索区间,因为通过IGV你会发现区间刚好存在一个breakpoint,所有双端联配在这里都出现不同程度的soft-clip。
samtools view -b -F 0x2 align/data.bwa.bam | samtools view -b -G 141 | samtools view -G 77 | cut -f 4 | sort | head -n2
samtools view -b -F 0x2 align/data.bwa.bam | samtools view -b -G 141 | samtools view -G 77 | cut -f 4 | sort | tail -n2
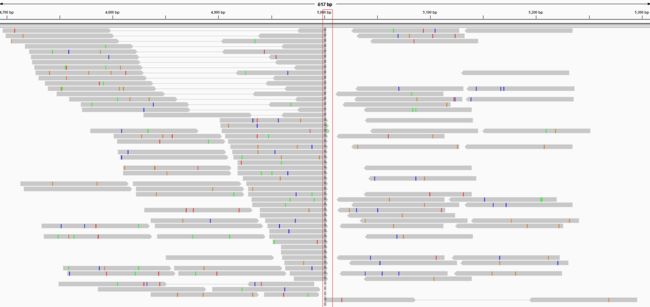
进一步寻找插入的具体位点可以有两种策略,一种是不知道插入的序列信息,一种是已知插入序列的序列信息。
未知序列信息
一种方案是将不完美比对序列进行组装,得到的较长序列通过BLASTN确定位置。组装可以使用velvet,用起来比较容易,而且可以用bioconda安装。
samtools view -b -F 0x2 align/data.bwa.bam | samtools sort -n | samtools fastq -1 read_1.fq -2 read_2.fq -
velveth velvet31 31 -fastq -separate -shortPaired read_1.fq read_2.fq
velvetg velvet31 -exp_cov auto -ins_length 350
# -ins_length表示插入文库大小
最后会在velvet31文件夹下生成contigs.fa,这里面的N50肯定是看不了的,我们只是需要一个比较长一点的序列而已。最后使用BLAST找到可能的位点。只需要建立索引数据库,然后用BLASTN去搜索组装的contigs.fa的可能位置。
cd refs
# 建立索引
makeblastdb -dbtype nucl -in ref.fa
# 搜索
blastn -query ../velvet31/contigs.fa -db ref.fa -outfmt 8
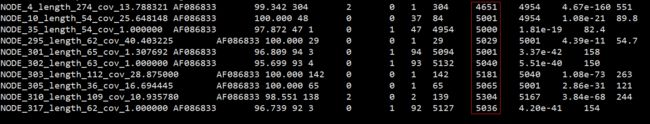
已知序列信息
如果序列已知,那么在找个基础上可以先按照未知序列信息的策略组装,只不过可以分别对T-DNA插入和参考基因组分别BLAST, 于是可以找到一个覆盖插入位点的contig。或者将这条插入序列加入参考序列中,从比对结果中过滤处配对的两个reads中,一个比对到原来的参考序列,一个比对到插入序列的结果。
建立索引并比对:
cat refs/AF086833.fa| seqret -filter -sbegin 5000 -send 8000 > refs/insertion.fas
bioawk -c fastx '{print ">insertion\n" $seq}' insertion.fas >> ref.fa
bwa index refs/ref.fa
bwa mem refs/ref.fa data/data.bwa.read1.fastq.gz data/data.bwa.read2.fastq.gz | samtools sort > align/data.bwa.new.bam
samtools index align/data.bwa.new.bam
从符合要求的序列中找到可能的插入位置
$ samtools view -f 0x2 align/data.bwa.new.bam | awk '{if($7!="=") print $0}' | cut -f 3,4
AF086833 4940
AF086833 4951
...
AF086833 5003
AF086833 5003
AF086833 5006