目录
R语言之生信①差异基因分析1
R语言之生信②差异基因分析2
R语言之生信③差异基因分析3
R语言之生信④TCGA生存分析1
R语言之生信⑤TCGA生存分析2
R语言之生信⑥TCGA生存分析3
R语言之生信⑦Cox比例风险模型(单因素)
- 加载数据
setwd("D:\\diff")
# Reading in count data
files <- c("GSM1545535_10_6_5_11.txt", "GSM1545536_9_6_5_11.txt",
"GSM1545538_purep53.txt","GSM1545539_JMS8-2.txt",
"GSM1545540_JMS8-3.txt","GSM1545541_JMS8-4.txt",
"GSM1545542_JMS8-5.txt","GSM1545544_JMS9-P7c.txt",
"GSM1545545_JMS9-P8c.txt")
read.delim(files[1], nrow=5)
- 加载limma以及修改样本名
library(limma)
library(edgeR)
x <- readDGE(files, columns=c(1,3))
class(x)
dim(x)
# Organising sample information
samplenames <- substring(colnames(x), 12, nchar(colnames(x)))
#截取x从第十二位到末尾的名字
samplenames
-加载样本分组信息
colnames(x) <- samplenames
group <- as.factor(c("LP", "ML", "Basal", "Basal",
"ML", "LP", "Basal", "ML", "LP"))
x$samples$group <- group
lane <- as.factor(rep(c("L004","L006","L008"), c(3,4,2)))
x$samples$lane <- lane
x$samples
> x$samples
files group lib.size norm.factors lane
10_6_5_11 GSM1545535_10_6_5_11.txt LP 32863052 1 L004
9_6_5_11 GSM1545536_9_6_5_11.txt ML 35335491 1 L004
purep53 GSM1545538_purep53.txt Basal 57160817 1 L004
JMS8-2 GSM1545539_JMS8-2.txt Basal 51368625 1 L006
JMS8-3 GSM1545540_JMS8-3.txt ML 75795034 1 L006
JMS8-4 GSM1545541_JMS8-4.txt LP 60517657 1 L006
JMS8-5 GSM1545542_JMS8-5.txt Basal 55086324 1 L006
JMS9-P7c GSM1545544_JMS9-P7c.txt ML 21311068 1 L008
JMS9-P8c GSM1545545_JMS9-P8c.txt LP 19958838 1 L008
- 基因注释
这些基因信息可以使用特定的生物体包来检索,例如Mus.musculus8用于小鼠(或Homo.sapiens9用于人类)或biomaRt包,其连接Ensembl基因组数据库以执行基因注释。可以检索的信息类型包括gene symbols, gene names, chromosome names and locations, Entrez gene IDs, Refseq gene IDs and Ensembl gene IDs 等等。 biomaRt主要处理Ensembl基因ID,而Mus.musculus包含各种来源的信息,并允许用户在许多不同的基因ID之间进行选择作为关键。我们的数据集中可用的Entrez基因ID使用Mus.musculus软件包进行注释,以检索相关的基因符号和染色体信息。
library(Mus.musculus)
geneid <- rownames(x)
genes <- select(Mus.musculus, keys=geneid, columns=c("SYMBOL", "TXCHROM"),
keytype="ENTREZID")
dim(genes)
head(genes)
#. For simplicity we do the latter,
# keeping only the first occurrence of each gene ID.
genes <- genes[!duplicated(genes$ENTREZID),]
x$genes <- genes
x
- 数据预处理
从原始尺度转换
对于差异表达和相关分析,基因表达很少在原始计数水平上考虑,因为文库测序的深度更大会导致更高的计数。相反,通常的做法是将原始计数转换为可以解决这种库大小差异的规模。流行的转换包括每百万次计数(CPM),每百万次计数(log-CPM),每千克转录本的读数(RPKM)和每千万转录本的百万分率(FPKM)。
在我们的分析中,CPM和log-CPM转换经常使用,尽管它们没有考虑RPKM和FPKM值所做的特征长度差异。尽管可以使用RPKM和FPKM值,但CPM和log-CPM值可以单独使用计数矩阵计算,并且足以用于我们感兴趣的比较类型。假设条件之间的异构体使用没有差异差异表达分析着眼于条件之间的基因表达变化,而不是比较多个基因的表达或得出绝对表达水平的结论。换句话说,基因长度对于感兴趣的比较保持不变,任何观察到的差异都是条件变化的结果,而不是基因长度的变化。
这里使用edgeR中的cpm函数将原始计数转换为CPM和log-CPM值,其中对数转换使用先前计数为0.25来避免采用零对数。如果基因长度可用,则RPKM值与使用edgeR中的rpkm函数的CPM值一样容易计算。
cpm <- cpm(x)
lcpm <- cpm(x, log=TRUE)
- 去除低表达的基因
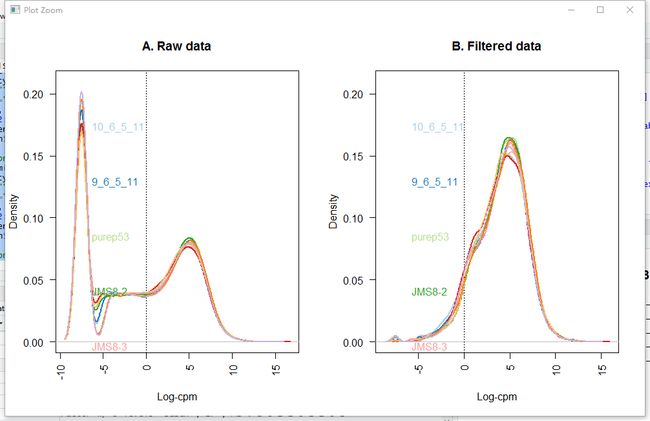
所有数据集将包括表达的基因和不表达的基因的组合。 虽然检查在一种条件下表达但不在另一种条件下表达的基因是有意义的,但是一些基因在所有样品中都未表达。 事实上,该数据集中19%的基因在所有九个样本中都有零个计数。
table(rowSums(x$counts==0)==9)
keep.exprs <- rowSums(cpm>1)>=3
x <- x[keep.exprs,, keep.lib.sizes=FALSE]
dim(x)
library(RColorBrewer)
nsamples <- ncol(x)
col <- brewer.pal(nsamples, "Paired")
par(mfrow=c(1,2))
plot(density(lcpm[,1]), col=col[1], lwd=2, ylim=c(0,0.21), las=2,
main="", xlab="")
title(main="A. Raw data", xlab="Log-cpm")
abline(v=0, lty=3)
for (i in 2:nsamples){
den <- density(lcpm[,i])
lines(den$x, den$y, col=col[i], lwd=2)
}
legend("topright", samplenames, text.col=col, bty="n")
lcpm <- cpm(x, log=TRUE)
plot(density(lcpm[,1]), col=col[1], lwd=2, ylim=c(0,0.21), las=2,
main="", xlab="")
title(main="B. Filtered data", xlab="Log-cpm")
abline(v=0, lty=3)
for (i in 2:nsamples){
den <- density(lcpm[,i])
lines(den$x, den$y, col=col[i], lwd=2)
}
legend("topright", samplenames, text.col=col, bty="n")
- 标准化基因表达分布
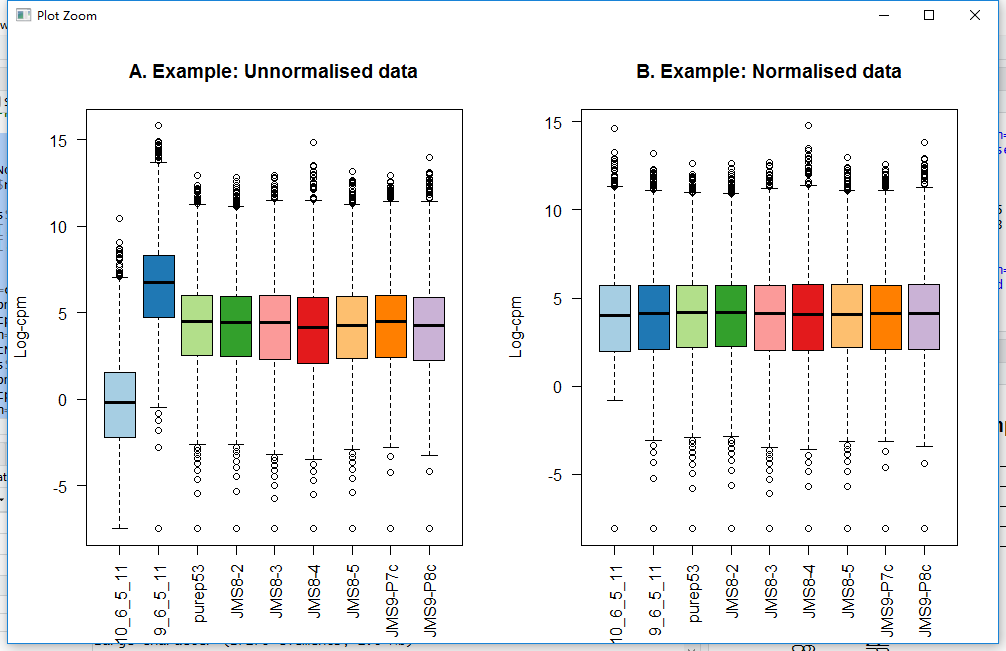
在样品制备或测序过程中,不具有生物学意义的外部因素会影响单个样品的表达。例如,与第二批处理的样品相比,在第一批实验中处理的样品可以具有更高的表达。假设所有样本应具有相似的表达值范围和分布。标准化需要确保每个样品的表达分布在整个实验中相似。
显示每个样本表达式分布(如密度或盒形图)的任何图都可用于确定是否有任何样本与其他样本不同。在这个数据集的所有样本中,log-CPM值的分布是相似的。
尽管如此,通过使用edgeR中的calcNormFactors函数来执行通过修剪M值平均值(TMM)的方法的归一化。这里计算的归一化因子用作库大小的缩放因子。使用DGEList对象时,这些规范化因子会自动存储在x norm.factors中。对于这个数据集,TMM归一化的影响是轻微的,这在比例因子的大小上是明显的,它们都相对接近于1。
x <- calcNormFactors(x, method = "TMM")
x$samples$norm.factors
x2 <- x
x2$samples$norm.factors <- 1
x2$counts[,1] <- ceiling(x2$counts[,1]*0.05)
x2$counts[,2] <- x2$counts[,2]*5
par(mfrow=c(1,2))
lcpm <- cpm(x2, log=TRUE)
boxplot(lcpm, las=2, col=col, main="")
title(main="A. Example: Unnormalised data",ylab="Log-cpm")
x2 <- calcNormFactors(x2)
x2$samples$norm.factors
lcpm <- cpm(x2, log=TRUE)
boxplot(lcpm, las=2, col=col, main="")
title(main="B. Example: Normalised data",ylab="Log-cpm")