网速慢的原因找到了

R 与 BioMart 数据库的语言接口 (interface)
Durinck S, Spellman P, Birney E, Huber W (2009). “Mapping identifiers for the integration of genomic datasets with the R/Bioconductor package biomaRt.”Nature Protocols, 4, 1184–1191.
Durinck S, Moreau Y, Kasprzyk A, Davis S, De Moor B, Brazma A, Huber W (2005). “BioMart and Bioconductor: a powerful link between biological databases and microarray data analysis.” Bioinformatics, 21, 3439–3440.
DOI: 10.18129/B9.bioc.biomaRt
1. 关于 biomaRt
biomaRt 向 R 和 BioMart software suite 的数据库(例如 Ensembl, Uniprot, HapMap)提供了一个接口,也就是通过R直接获取数据库中的信息。
查看可以连接到的 BioMart 数据库:
library(biomaRt)
listMarts()
# biomart version
# 1 ENSEMBL_MART_ENSEMBL Ensembl Genes 96
# 2 ENSEMBL_MART_MOUSE Mouse strains 96
# 3 ENSEMBL_MART_SNP Ensembl Variation 96
# 4 ENSEMBL_MART_FUNCGEN Ensembl Regulation 96
2. 选择数据库及数据集
通过函数 useMart 设置要连接的 BioMart 数据集。
ensembl = useMart("ensembl")
对于 Ensembl , 每一个物种又是一个独立的数据集,需要用到 listDatasets() 和 useDataset() 查看和启用数据集。
head(listDatasets(ensembl))
# dataset
# 1 abrachyrhynchus_gene_ensembl
# 2 acalliptera_gene_ensembl
# 3 acarolinensis_gene_ensembl
# 4 acitrinellus_gene_ensembl
# 5 ahaastii_gene_ensembl
# 6 amelanoleuca_gene_ensembl
# description version
# 1 Pink-footed goose genes (ASM259213v1) ASM259213v1
# 2 Eastern happy genes (fAstCal1.2) fAstCal1.2
# 3 Anole lizard genes (AnoCar2.0) AnoCar2.0
# 4 Midas cichlid genes (Midas_v5) Midas_v5
# 5 Great spotted kiwi genes (aptHaa1) aptHaa1
# 6 Panda genes (ailMel1) ailMel1
ensembl = useDataset("hsapiens_gene_ensembl",mart=ensembl)
如果已经确定了要用的数据集,可以这样一步设置:
ensembl = useMart(biomart = "ensembl",
dataset = "hsapiens_gene_ensembl")
## "biomart="和"dataset="可以省略
3. 设置各类参数,建立检索
getBM() 是检索数据用到的主要函数,首先需要对它的4个参数 (filters, attributes, values, mart) 及参数选项进行查看和选择。
-
filters, input
head(listFilters(ensembl)) # name description # 1 chromosome_name Chromosome/scaffold name # 2 start Start # 3 end End # 4 band_start Band Start # 5 band_end Band End # 6 marker_start Marker Start -
attributes, output
head(listAttributes(ensembl)) # name description # 1 ensembl_gene_id Gene stable ID # 2 ensembl_gene_id_version Gene stable ID version # 3 ensembl_transcript_id Transcript stable ID # 4 ensembl_transcript_id_version Transcript stable ID version # 5 ensembl_peptide_id Protein stable ID # 6 ensembl_peptide_id_version Protein stable ID version # page # 1 feature_page # 2 feature_page # 3 feature_page # 4 feature_page # 5 feature_page # 6 feature_page -
values
应用于 "filters" 的值,用于检索。
-
mart
已选择的由
useMart()得到的对象。
## 搜索可用选项
searchAttributes(mart = ensembl,pattern = "")
searchFilters(mart = ensembl, pattern = "")
listFilterValues(mart = ensembl, filter = "")
4. biomaRt 检索实例
4.1 Affymetrix id → HGNC symbol + 染色体名 + 染色体坐标
已知 Affymetrix hgu133plus2 id,想得到 HGNC symbol, 染色体名、染色体坐标 (start and end positions).
比如要搜索含 ”affy“ 的 attributes :
searchAttributes(mart = ensembl,pattern = "affy")
# name description page
# 96 affy_hc_g110 AFFY HC G110 probe feature_page
# 97 affy_hg_focus AFFY HG Focus probe feature_page
# 98 affy_hg_u133a AFFY HG U133A probe feature_page
# 99 affy_hg_u133a_2 AFFY HG U133A 2 probe feature_page
# 100 affy_hg_u133b AFFY HG U133B probe feature_page
# 101 affy_hg_u133_plus_2 AFFY HG U133 Plus 2 probe feature_page
# 102 affy_hg_u95a AFFY HG U95A probe feature_page
# 103 affy_hg_u95av2 AFFY HG U95Av2 probe feature_page
# 104 affy_hg_u95b AFFY HG U95B probe feature_page
# 105 affy_hg_u95c AFFY HG U95C probe feature_page
# 106 affy_hg_u95d AFFY HG U95D probe feature_page
# 107 affy_hg_u95e AFFY HG U95E probe feature_page
# 108 affy_hta_2_0 AFFY HTA 2 0 probe feature_page
# 109 affy_huex_1_0_st_v2 AFFY HuEx 1 0 st v2 probe feature_page
# 110 affy_hugenefl AFFY HuGeneFL probe feature_page
# 111 affy_hugene_1_0_st_v1 AFFY HuGene 1 0 st v1 probe feature_page
# 112 affy_hugene_2_0_st_v1 AFFY HuGene 2 0 st v1 probe feature_page
# 113 affy_primeview AFFY PrimeView probe feature_page
# 114 affy_u133_x3p AFFY U133 X3P probe feature_page
也可以这样:
attributes <- listAttributes(ensembl)
attributes[grep("affy", attributes$name),]
## 得到和上面一样的结果
下面是正文
affyids = c("202763_at","209310_s_at","207500_at")
getBM(attributes = c('affy_hg_u133_plus_2', 'hgnc_symbol', 'chromosome_name',
'start_position', 'end_position', 'band'),
filters = 'affy_hg_u133_plus_2',
values = affyids,
mart = ensembl)
# affy_hg_u133_plus_2 hgnc_symbol chromosome_name
# 1 202763_at CASP3 4
# 2 209310_s_at CASP4 11
# 3 207500_at CASP5 11
# start_position end_position band
# 1 184627696 184649509 q35.1
# 2 104942866 104969366 q22.3
# 3 104994235 105023168 q22.3
4.2 EntrezGene id → GO 注释
已知 entrez id, 想得到对应的 GO id.
entrez = c("673","837")
goids = getBM(attributes = c('entrezgene', 'go_id'),
filters = 'entrezgene',
values = entrez,
mart = ensembl)
head(goids)
# entrezgene go_id
# 1 673 GO:0005524
# 2 673 GO:0007165
# 3 673 GO:0006468
# 4 673 GO:0035556
# 5 673 GO:0004672
# 6 673 GO:0046872
作者在这里其实是想得到 "GO identifiers related to biological processes", 然鹅并没有看出来是如何把 GO id 限定在 BP 范围的...比如第一个就不是。

4.3 GO term + 染色体名 → HGNC symbol
已知 GO id 和染色体名, 想得到对应的所有基因的 HGNC symbol.
go = c("GO:0051330","GO:0000080","GO:0000114","GO:0000082")
chrom = c(17,20,"Y")
getBM(attributes = "hgnc_symbol",
filters = c("go","chromosome_name"),
values = list(go, chrom),
mart = ensembl) ## 有多个filter时,'values'应为 list
# hgnc_symbol
# 1 RPS6KB1
# 2 RPA1
# 3 CDK3
# 4 CDC6
# 5 MCM8
# 6 CRLF3
4.4 RefSeq id → 蛋白质结构域 id + 描述
已知 RefSeq id, 注释 INTERPRO 蛋白质结构域 id 及描述
refseqids = c("NM_005359","NM_000546")
getBM(attributes = c("refseq_mrna","interpro",
"interpro_description"),
filters = "refseq_mrna",
values = refseqids,
mart = ensembl)
# refseq_mrna interpro
# 1 NM_000546 IPR002117
# 2 NM_000546 IPR008967
# 3 NM_000546 IPR010991
# 4 NM_000546 IPR011615
# 5 NM_000546 IPR012346
# 6 NM_000546 IPR013872
# 7 NM_000546 IPR036674
# 8 NM_005359 IPR001132
# 9 NM_005359 IPR003619
# 10 NM_005359 IPR008984
# 11 NM_005359 IPR013019
# 12 NM_005359 IPR013790
# 13 NM_005359 IPR017855
# 14 NM_005359 IPR036578
# interpro_description
# 1 p53 tumour suppressor family
# 2 p53-like transcription factor, DNA-binding
# 3 p53, tetramerisation domain
# 4 p53, DNA-binding domain
# 5 p53/RUNT-type transcription factor, DNA-binding domain superfamily
# 6 p53 transactivation domain
# 7 p53-like tetramerisation domain superfamily
# 8 SMAD domain, Dwarfin-type
# 9 MAD homology 1, Dwarfin-type
# 10 SMAD/FHA domain superfamily
# 11 MAD homology, MH1
# 12 Dwarfin
# 13 SMAD-like domain superfamily
# 14 SMAD MH1 domain superfamily
4.5 染色体名 + 坐标 → Affymetrix id + Ensembl id
已知染色体名和碱基区间,想得到区间内基因的 hgu133plus2 芯片的 Affymetrix id 及 Ensembl id.
getBM(attributes = c('affy_hg_u133_plus_2','ensembl_gene_id'),
filters = c('chromosome_name','start','end'),
values = list(16,1100000,1250000),
mart = ensembl)
# affy_hg_u133_plus_2 ensembl_gene_id
# 1 ENSG00000260702
# 2 215502_at ENSG00000260532
# 3 ENSG00000273551
# 4 205845_at ENSG00000196557
# 5 ENSG00000196557
# 6 ENSG00000260403
# 7 ENSG00000259910
# 8 ENSG00000261294
# 9 220339_s_at ENSG00000116176
# 10 ENSG00000277010
# 11 205683_x_at ENSG00000197253
# 12 207134_x_at ENSG00000197253
# 13 217023_x_at ENSG00000197253
# 14 210084_x_at ENSG00000197253
# 15 215382_x_at ENSG00000197253
# 16 216474_x_at ENSG00000197253
# 17 205683_x_at ENSG00000172236
# 18 207134_x_at ENSG00000172236
# 19 217023_x_at ENSG00000172236
# 20 210084_x_at ENSG00000172236
# 21 215382_x_at ENSG00000172236
# 22 216474_x_at ENSG00000172236
4.6 GO id → entrezgene id + HGNC symbol
已知 GO id, 想得到对应所有基因的 entrezgene id 和 HGNC symbol。
getBM(attributes = c('entrezgene','hgnc_symbol'),
filters = 'go',
values = 'GO:0004707',
mart = ensembl)
# entrezgene hgnc_symbol
# 1 5602 MAPK10
# 2 5594 MAPK1
# 3 5597 MAPK6
# 4 5599 MAPK8
# 5 225689 MAPK15
# 6 6300 MAPK12
# 7 5595 MAPK3
# 8 5600 MAPK11
# 9 5598 MAPK7
# 10 5596 MAPK4
# 11 51701 NLK
# 12 5601 MAPK9
# 13 1432 MAPK14
# 14 5603 MAPK13
4.7 EntrezGene id → 启动子序列
函数 getSequence() 可以通过染色体坐标或 id 获取序列。type 可以通过 listFilters() 查询。
seqType可以有的选择:cdna, peptide, 3utr, 5utr, gene_exon, transcript_exon, transcript_exon_intron, gene_exon_intron, coding, coding_transcript_flank, coding_gene_flank, transcript_flank, gene_flank, coding_gene_flank
entrez = c("673","7157","837")
getSequence(id = entrez,
type = "entrezgene",
seqType = "coding_gene_flank",
upstream = 100,
mart = ensembl)
# coding_gene_flank
# 1 CACGTTTCCGCCCTTTGCAATAAGGAAATACATAGTTTACTTTCATTTTTGACTCTGAGGCTCTTTCCAACGCTGTAAAAAAGGACAGAGGCTGTTCCCT
# 2 TCCTTCTCTGCAGGCCCAGGTGACCCAGGGTTGGAAGTGTCTCATGCTGGATCCCCACTTTTCCTCTTGCAGCAGCCAGACTGCCTTCCGGGTCACTGCC
# 3 CCTCCGCCTCCGCCTCCGCCTCCGCCTCCCCCAGCTCTCCGCCTCCCTTCCCCCTCCCCGCCCGACAGCGGCCGCTCGGGCCCCGGCTCTCGGTTATAAG
# entrezgene
# 1 837
# 2 7157
# 3 673
4.8 染色体坐标 → 5' UTR 序列
通过染色体坐标用 getSequence() 获取区间内所有基因的 5' UTR 序列,并显示entrezgene id.
getSequence(chromosome = 3, start = 185514033, end = 185535839,
type = "entrezgene",
seqType = "5utr",
mart = ensembl)
# # 5utr
# 1 Sequence unavailable
# 2 ACCACACCTCTGAGTCGTCTGAGCTCACTGTGAGCAAAATCCCACAGTGGAAACTCTTAAGCCTCTGCGAAGTAAATCATTCTTGTGAATGTGACACACGATCTCTCCAGTTTCCAT
# 3 TGAGCAAAATCCCACAGTGGAAACTCTTAAGCCTCTGCGAAGTAAATCATTCTTGTGAATGTGACACACGATCTCTCCAGTTTCCAT
# 4 ATTCTTGTGAATGTGACACACGATCTCTCCAGTTTCCAT
# entrezgene
# 1 200879
# 2 200879
# 3 200879
# 4 200879
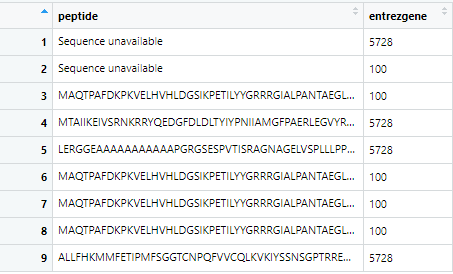
4.9 EntrezGene id → 蛋白质序列
entrez = c("100","5728")
protein = getSequence(id = entrez,
type = "entrezgene",
seqType = "peptide",
mart = ensembl)
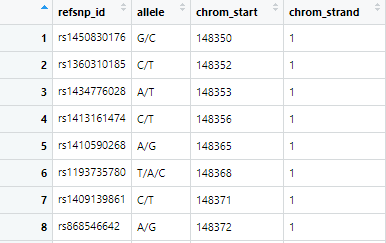
4.10 染色体坐标 → SNPs
连接 SNP 数据库:
snpmart = useMart(biomart = "ENSEMBL_MART_SNP",
dataset = "hsapiens_snp")
SNPs = getBM(attributes = c('refsnp_id','allele',
'chrom_start','chrom_strand'),
filters = c('chr_name','start','end'),
values = list(8,148350,148612),
mart = snpmart)
nrow(SNPs)
# [1] 68
4.11 Gene symbol → 染色体坐标 + 同源基因的染色体坐标 + RefSeq
已知基因名,获取该基因在人类染色体上的坐标, 和其小鼠同源基因的染色体坐标及 RefSeq id.
函数 getLDS() (Get Linked Dataset) 可连接两个不同的 BioMart 数据集并建立检索。
human = useMart("ensembl", dataset = "hsapiens_gene_ensembl")
mouse = useMart("ensembl", dataset = "mmusculus_gene_ensembl")
getLDS(attributes = c("hgnc_symbol","chromosome_name", "start_position"),
filters = "hgnc_symbol",
values = "TP53",
mart = human,
attributesL = c("refseq_mrna","chromosome_name","start_position"),
martL = mouse) ## martL, Mart object representing linked dataset
# HGNC.symbol Chromosome.scaffold.name Gene.start..bp. RefSeq.mRNA.ID
# 1 TP53 17 7661779
# 2 TP53 17 7661779 NM_001127233
# 3 TP53 17 7661779 NM_011640
# Chromosome.scaffold.name.1 Gene.start..bp..1
# 1 11 69580359
# 2 11 69580359
# 3 11 69580359
5. 输出 FASTA 文件
函数 exportFASTA() 可以将 getSequence() 的结果输出为 FASTA 文件。
utr5seq = getSequence(chromosome=3, start=185514033, end=185535839,
type="entrezgene",
seqType="5utr",
mart=ensembl)
exportFASTA(utr5seq,"5UTRseq.fasta")
6. 更细致的检索方法
select(), columns(), keytypes(), keys() . 这些函数和 getBM() 类似,但组合使用能够使检索过程更便捷。
head(columns(ensembl))
# [1] "3_utr_end" "3_utr_end" "3_utr_start" "3_utr_start" "3utr"
# [6] "5_utr_end"
head(keytypes(ensembl))
# [1] "affy_hc_g110" "affy_hg_focus" "affy_hg_u133_plus_2"
# [4] "affy_hg_u133a" "affy_hg_u133a_2" "affy_hg_u133b"
affy=c("202763_at","209310_s_at","207500_at")
select(ensembl, keys=affy, columns=c('affy_hg_u133_plus_2','entrezgene'),
keytype='affy_hg_u133_plus_2')
# affy_hg_u133_plus_2 entrezgene
# 1 202763_at 836
# 2 209310_s_at 837
# 3 207500_at 838
References
-
The biomaRt users guide
https://bioc.ism.ac.jp/packages/devel/bioc/vignettes/biomaRt/inst/doc/biomaRt.html#how-to-build-a-biomart-query>
-
Some basics of biomaRt
https://www.r-bloggers.com/some-basics-of-biomart/
*悄咪咪吐槽
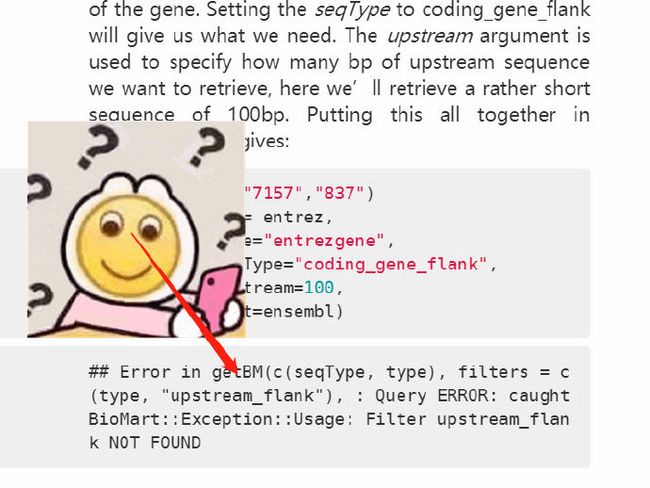

都是为什么呢。
最后,向大家隆重推荐生信技能树的一系列干货!
- 生信技能树全球公益巡讲:https://mp.weixin.qq.com/s/E9ykuIbc-2Ja9HOY0bn_6g
- B站公益74小时生信工程师教学视频合辑:https://mp.weixin.qq.com/s/IyFK7l_WBAiUgqQi8O7Hxw
- 招学徒:https://mp.weixin.qq.com/s/KgbilzXnFjbKKunuw7NVfw