在实战之前,首先对CHIP-seq分析做一些了解,以下是从各个地方copy过来综合起来的,有些散乱,是我认为重要以及应该了解的知识点。
chip-seq 实战就跟在转录组和外显子上的实战是没有本质区别的,只是它多了一些分析,转录组只找表达量做差异分析,外显子只找变异做变异注释,那chip-seq 找的是peaks和motif 。
这里biobabble公众号有比较完整的chip-seq的介绍:
CS1: ChIPseq简介
CS2: BED文件
CS3: peak注释
CS4:关于ChIPseq注释的几个问题
CS5: 吃着火锅,唱着歌,还把分析给做了
CS6: ChIPseeker的可视化方法
CS7:Genomic coordination的富集性分析(1)
CS8:Genomic coordination的富集性分析(2)
CS9: GEO数据挖掘
CS番外1:如何定义下游的区间?
ChIP-seq 数据分析流程
1.得到原始序列文件后,第一步是执行标准的高通量数据质量控制。
2.原始的 reads 回比到研究物种的参考基因组上
3.特异性 ChIP-seq 的质量控制,检查 ChIP-seq 样品中的富集情况,并排除过度碎片
4.【核心】鉴定全基因上感兴趣的因子富集的区域,也叫 Peak calling
5.一旦确定了峰值区域,还可以评估需要峰值位置的特定 ChIP-seq 的质量控制措施
6.在 Peak calling 之后,要确保预测的峰值准确地捕捉到binding pattern。
7.Peak calling后的分析取决于实验设计与生物学问题。比如,如果在实验中有重复和/或多个条件,那么下一步可能是样本之间的比较(见第 8 章)。生物学重复将提供有关数据(噪声)中的重现性和内在生物和技术变异性的信息。差异结合分析解决了哪一Peak区域在两种情况下显示出明显不同的丰度(例如:与观察到的相同条件的重复之间的变化相比,差异比预期的要大得多)。
8.Peak区间或差异Peak做下游分析,比如基因组注释、GO分析、Pathway 分析、motif 查找、与其他基因组数据联合分析。(参考文章:https://www.jianshu.com/p/19b178ea406e)
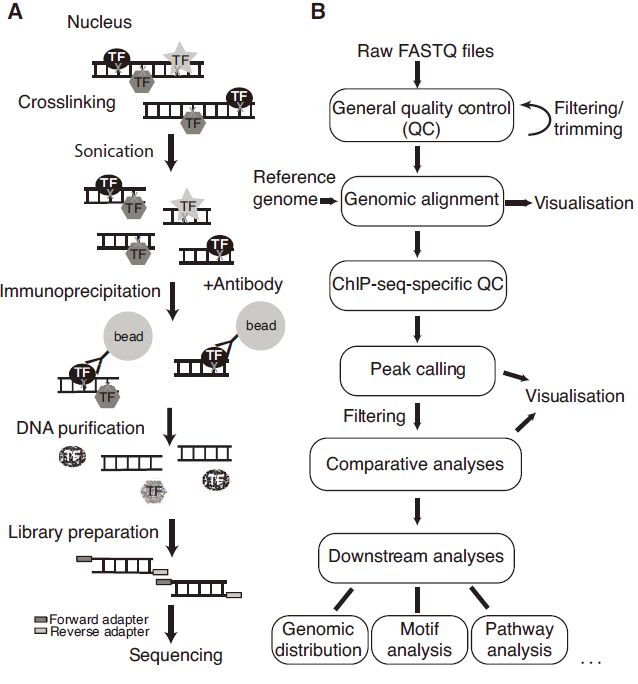
怎样识别富集区域?(https://www.jianshu.com/p/df33bcb68548)
从DNA测序的角度来说,因为测序都是5'端的reads,对于一个DNA序列来说(有正负链的),它mapping的位置正负链都有的(也就是红色和蓝色的reads都有),对这些reads位置进行统计画图可以看到一个红色的peak,一个蓝色的peak。这两个peak说明的是一个事情,就是这个地方有富集。最后对这两个peak进行merge,最后变成了一个富集区域。灰色的peak!
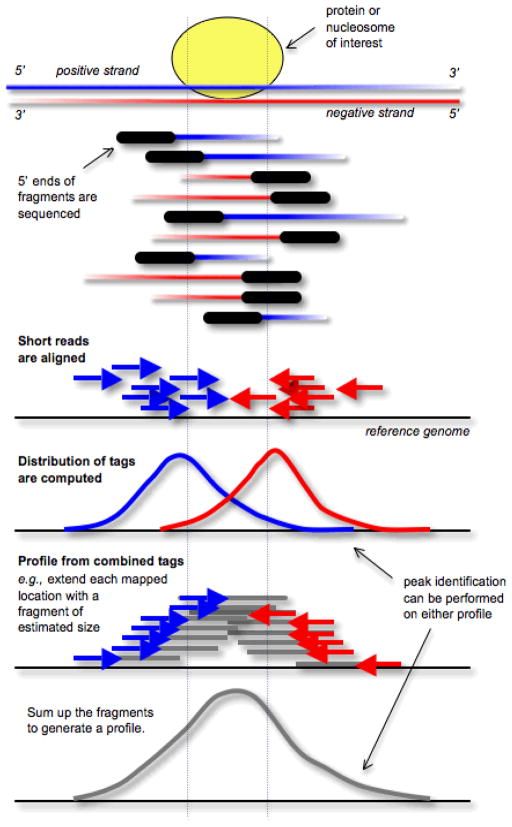
ChIP-seq的常见峰型
不同类型的蛋白或者组蛋白修饰会得到不同的峰形:
sharp binding sites, CTCF (red);
a mixture of shapes, RNA Polymerase II (orange);
medium size, H3K36me3 (green);
large domains, H3K27me3 (blue)
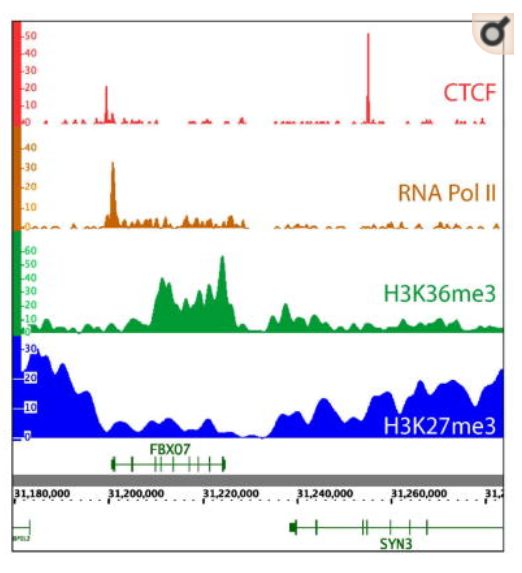
富集倍数
一般来说富集倍数要在5以上才算是显著,如何计算富集比率呢?看图~
Chip-Seq实践
需要用的软件:
fastqc, sra-tools, bowtie2, samtools, MACS2, deeptools
上述软件之前在RNA-seq实践中已经安装了大部分,需要安装MACS2和deeptools。
MACS2比较特殊,因为它需要Python2.7的环境才能安装,而我的电脑安装的是python3环境。所以首先创建一个虚拟环境,在python2.7的虚拟环境中使用(https://mp.weixin.qq.com/s/mEwl7-f2WTNfmK-FkNkP0A):
conda create -n py27 python=2
source activate py27
pip install MACS2
本次实践的文章是:
RYBP and Cbx7 define specific biological functions of polycomb complexes in mouse embryonic stem cells
(探索PRC1,PCR2蛋白复合物,不是转录因子或者组蛋白的CHIP-seq)
本文主要讲了PRC1(Polycomb repressive complex 1)在小鼠的胚胎干细胞中有两类亚型Cbx7-PRC1和RYBP-PRC1,但是Cbx7和RYBP这两种是不能共存的,尽管两者的全基因组定位在某些gene上交叉,也就是说在基因组上结合在了同一个位置。在分子水平上,Cbx7用于招募Ring1B结合到染色质上,然而RYBP可以增强PRC1的酶活性。RYBP结合的基因,其有着了较低水平的Ring1B和H2AK119ub,但其相比结合Cbx7有较高的表达量。在功能上,RYBP结合的基因主要涉及代谢调节和细胞周期,Cbx7结合的基因主要涉及early-lineage commitment of ESCs。(这段来源:http://www.bioinfo-scrounger.com/archives/358)
1.下载原始数据:
可以写一个脚本,批量下载:
#!/bin/bash
for ((i=204;i<=209;i++))
do wget https://sra-download.ncbi.nlm.nih.gov/traces/sra6/SRR/000605/SRR620$i
done
2.下载之后提取fastq文件:
#!/bin/bash
for i in SRR62020*
do
echo $i
fastq-dump --gzip --split-files $i
done
3.fastqc检查测序质量
fastqc发现Ring1B、Suz12,IgG_old,IgG样本的3’端有大约5个bp长度的碱基质量不是太好,可以在之后的bowtie2比对的时候设置参数,把质量差的几个碱基去掉。
4.bowtie2比对
首先下载bowtie2的mm10基因组索引:
wget ftp://ftp.ccb.jhu.edu/pub/data/bowtie2_indexes/mm10.zip
还需要mm10 refseq注释bed文件,进行网址下载http://genome.ucsc.edu/cgi-bin/hgTables,其中track和table选择RefSeq,然后输出格式为bed即可,最后下载(可以先在自己电脑上下载再传到服务器上,也可以像下面直接在服务器上下载)(参考http://www.bioinfo-scrounger.com/archives/358)
curl 'http://genome.ucsc.edu/cgi-bin/hgTables?hgsid=646311755_P0RcOBvAQnWZSzQz2fQfBiPPSBen&boolshad.hgta_printCustomTrackHeaders=0&hgta_ctName=tb_ncbiRefSeq&hgta_ctDesc=table+browser+query+on+ncbiRefSeq&hgta_ctVis=pack&hgta_ctUrl=&fbQual=whole&fbUpBases=200&fbExonBases=0&fbIntronBases=0&fbDownBases=200&hgta_doGetBed=get+BED' >mm10.refseq.bed
用bowtie2进行比对(用脚本进行):
#!/bin/bash
bowtie2 -p 6 -3 5 --local -x /media/yanfang/FYWD/CHIPSEQ/bowtie2mm10/mm10 -U /media/yanfang/FYWD/CHIPSEQ/fqfile/SRR620204_1.fastq.gz | samtools sort -O bam -o ring1B.bam
bowtie2 -p 6 -3 5 --local -x /media/yanfang/FYWD/CHIPSEQ/bowtie2mm10/mm10 -U /media/yanfang/FYWD/CHIPSEQ/fqfile/SRR620205_1.fastq.gz | samtools sort -O bam -o cbx7.bam
bowtie2 -p 6 -3 5 --local -x /media/yanfang/FYWD/CHIPSEQ/bowtie2mm10/mm10 -U /media/yanfang/FYWD/CHIPSEQ/fqfile/SRR620206_1.fastq.gz | samtools sort -O bam -o Suz12.bam
bowtie2 -p 6 -3 5 --local -x /media/yanfang/FYWD/CHIPSEQ/bowtie2mm10/mm10 -U /media/yanfang/FYWD/CHIPSEQ/fqfile/SRR620207_1.fastq.gz | samtools sort -O bam -o RYBP.bam
bowtie2 -p 6 -3 5 --local -x /media/yanfang/FYWD/CHIPSEQ/bowtie2mm10/mm10 -U /media/yanfang/FYWD/CHIPSEQ/fqfile/SRR620208_1.fastq.gz | samtools sort -O bam -o IgGold.bam
bowtie2 -p 6 -3 5 --local -x /media/yanfang/FYWD/CHIPSEQ/bowtie2mm10/mm10 -U /media/yanfang/FYWD/CHIPSEQ/fqfile/SRR620209_1.fastq.gz | samtools sort -O bam -o IgG.bam
~
但是貌似对比率不高:
19687027 reads; of these:
19687027 (100.00%) were unpaired; of these:
7778437 (39.51%) aligned 0 times
7766495 (39.45%) aligned exactly 1 time
4142095 (21.04%) aligned >1 times
60.49% overall alignment rate
[bam_sort_core] merging from 4 files and 1 in-memory blocks...
20710168 reads; of these:
20710168 (100.00%) were unpaired; of these:
2584870 (12.48%) aligned 0 times
10914615 (52.70%) aligned exactly 1 time
7210683 (34.82%) aligned >1 times
87.52% overall alignment rate
[bam_sort_core] merging from 4 files and 1 in-memory blocks...
21551927 reads; of these:
21551927 (100.00%) were unpaired; of these:
7109852 (32.99%) aligned 0 times
9455003 (43.87%) aligned exactly 1 time
4987072 (23.14%) aligned >1 times
67.01% overall alignment rate
[bam_sort_core] merging from 4 files and 1 in-memory blocks...
39870646 reads; of these:
39870646 (100.00%) were unpaired; of these:
6312903 (15.83%) aligned 0 times
20702502 (51.92%) aligned exactly 1 time
12855241 (32.24%) aligned >1 times
84.17% overall alignment rate
[bam_sort_core] merging from 8 files and 1 in-memory blocks...
13291429 reads; of these:
13291429 (100.00%) were unpaired; of these:
5608796 (42.20%) aligned 0 times
4663584 (35.09%) aligned exactly 1 time
3019049 (22.71%) aligned >1 times
57.80% overall alignment rate
[bam_sort_core] merging from 2 files and 1 in-memory blocks...
31218866 reads; of these:
31218866 (100.00%) were unpaired; of these:
5370101 (17.20%) aligned 0 times
15180536 (48.63%) aligned exactly 1 time
10668229 (34.17%) aligned >1 times
82.80% overall alignment rate
[bam_sort_core] merging from 6 files and 1 in-memory blocks...
5.用MACS2进行call peak:
首先要进入python2.7的环境:
source activate py27
然后写一个脚本:
#!/bin/bash
macs2 callpeak -c /media/yanfang/FYWD/CHIPSEQ/sambam/IgGold.bam -t /media/yanfang/FYWD/CHIPSEQ/sambam/Suz12.bam -m 10 30 -p 1e-5 -f BAM -g mm -n Suz12 2>Suz12.masc2.log
macs2 callpeak -c /media/yanfang/FYWD/CHIPSEQ/sambam/IgGold.bam -t /media/yanfang/FYWD/CHIPSEQ/sambam/ring1B.bam -m 10 30 -p 1e-5 -f BAM -g mm -n ring1B 2>ring1B.masc2.log
macs2 callpeak -c /media/yanfang/FYWD/CHIPSEQ/sambam/IgGold.bam -t /media/yanfang/FYWD/CHIPSEQ/sambam/cbx7.bam -m 10 30 -p 1e-5 -f BAM -g mm -n cxb7 2>cxb7.masc2.log
macs2 callpeak -c /media/yanfang/FYWD/CHIPSEQ/sambam/IgGold.bam -t /media/yanfang/FYWD/CHIPSEQ/sambam/RYBP.bam -m 10 30 -p 1e-5 -f BAM -g mm -n RYBP 2>RYBP.masc2.log
-t/–treatment 输入文件,支持很多格式,搭配-f/–format使用
-f/–format 设定输入文件的格式,这里选择bam格式
-c/–control 对照组(或者模拟的)数据,需要跟-t的输出文件在相同目录下
-n/–name 输出文件(有很多个文件)的前缀
–outdir 输出文件所在目录(不设定的话就默认当前目录了)
-g/–gsize 提供基因组的大小,程序有默认的几个物种可以选hs,mm,ce,dm
-q/–qvalue 设定FDR的阈值,默认是0.05
-p/–pvalue 设定pvalue的阈值,如果参数设定了-p,则其会覆盖参数-q的作用
-B/–bdg If this flag is on, MACS will store the fragment pileup, control lambda, -log10pvalue and -log10qvalue scores in bedGraph files.
参考文章(http://www.bioinfo-scrounger.com/archives/363)
运行后得到的文件不只是上述提到的一类,还有如下格式:
1.NAME_peaks.xls: 以表格形式存放peak信息,虽然后缀是xls,但其实能用文本编辑器打开,和bed格式类似,但是以1为基,而bed文件是以0为基.也就是说xls的坐标都要减一才是bed文件的坐标。
2.NAME_peaks.narrowPeak: NAME_peaks.broadPeak 类似。后面4列表示为, integer score for display, fold-change,-log10 pvalue,-log10 qvalue,relative summit position to peak start。内容和NAME_peaks.xls基本一致,适合用于导入R进行分析。
3.NAME_summits.bed:记录每个peak的peak summits,话句话说就是记录极值点的位置。MACS建议用该文件寻找结合位点的motif。
4.NAME_model.r: 能通过$ Rscript NAME_model.r作图,得到是基于你提供数据的peak模型。
运行之后,看一下每个样本找到多少个peak(bed的格式,储存了每个peaks的位置信息):
$ wc -l *.bed
2754 cxb7_summits.bed
7193 ring1B_summits.bed
473 RYBP_summits.bed
6696 Suz12_summits.bed
本文作者在GEO给的PEAKS个数如下:
2754 GSE42466_Cbx7_peaks_10.txt
6982 GSE42466_Ring1b_peaks_10.txt
6872 GSE42466_RYBP_peaks_5.txt
8054 GSE42466_Suz12_peaks_10.txt
然而我查了几篇对该文献重复的文章,call peak的数目和文章也有较大差距。(https://cloud.tencent.com/developer/article/1055109
http://www.bioinfo-scrounger.com/archives/363
https://mp.weixin.qq.com/s/_A0rHldzEgVk7bgwt457qQ?
https://mp.weixin.qq.com/s/mEwl7-f2WTNfmK-FkNkP0A)
6.结果注释与可视化
可视化这里我查阅了很多文章,分成两种,有些人用的是chpseeker包进行peak的可视化和注释;而还有一些人用了deeptool软件进行分析。我两种都试了试。
首先我用的是Chipseeker包。
ChIPseeker的功能分为三类(https://www.jianshu.com/p/c83a38915afc):
(1) 注释:提取peak附近最近的基因, 注释peak所在区域
(2)比较:估计ChIP peak数据集中重叠部分的显著性;整合GEO数据集,以便于将当前结果和已知结果比较
(3)可视化: peak的覆盖情况;TSS区域结合的peak的平均表达谱和热图;基因组注释;TSS距离;peak和基因的重叠。
Chip peaks coverage plot
#调用要用的包
library("ChIPseeker")
library("org.Mm.eg.db")
library("TxDb.Mmusculus.UCSC.mm10.knownGene")
library("clusterProfiler")
#定位文件位置
setwd("/media/yanfang/FYWD/CHIPSEQ/bed")
#读入bed文件
cbx7 <- readPeakFile("cxb7_peaks.narrowPeak.bed")
ring1B <- readPeakFile("ring1B_peaks.narrowPeak.bed")
Suz12 <- readPeakFile("Suz12_peaks.narrowPeak.bed")
#查看不同的蛋白的peak在染色体上的分布,因为RYBP的peak几乎没有,所以没有用这个蛋白做后续的画图处理
covplot(cbx7, weightCol="V5")
covplot(ring1B, weightCol="V5")
covplot(Suz12, weightCol="V5")

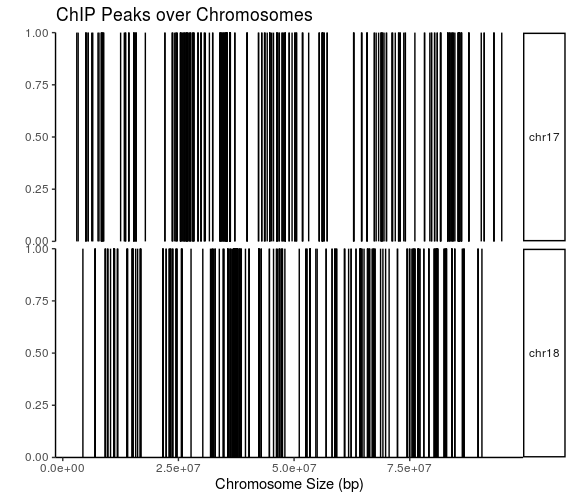
#查看在指定的染色体上的分布
covplot(cbx7,chrs=c("chr17", "chr18"))
热图(Heatmap of ChIP binding to TSS regions)
# 定义TSS上下游的距离
> promoter <- promoters(TxDb.Mmusculus.UCSC.mm10.knownGene, upstream = 3000, downstream = 3000)
> list_peak <- list(cbx7, ring1B, Suz12)
> tagMatrix <- lapply(list_peak, getTagMatrix, window=promoter)
> tagHeatmap(tagMatrix, xlim=c(-3000, 3000), color="red")

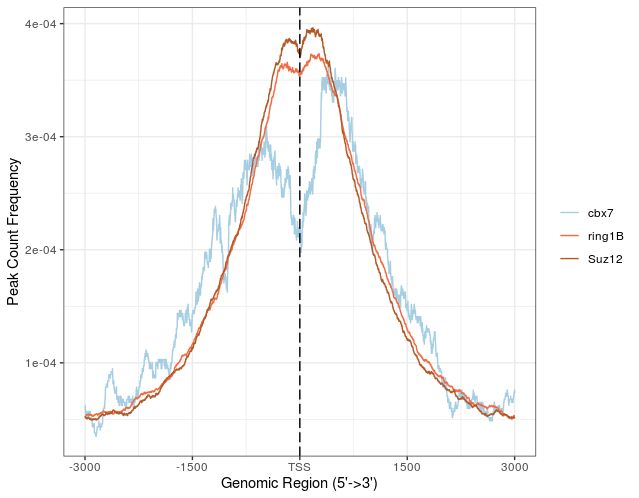
Average Profile of ChIP peaks binding to TSS region
用谱图的形式来展示不同蛋白结合的强度:
> library(org.Mm.eg.db)
> library(TxDb.Mmusculus.UCSC.mm10.knownGene)
> library(VennDiagram)
> setwd("/media/yanfang/FYWD/CHIPSEQ/bed")
> cbx7 <- readPeakFile("cxb7_peaks.narrowPeak.bed")
> ring1B <- readPeakFile("ring1B_peaks.narrowPeak.bed")
> Suz12 <- readPeakFile("Suz12_peaks.narrowPeak.bed")
> peaks <- list(cbx7=cbx7,ring1B=ring1B,Suz12=Suz12)
> txdb <- TxDb.Mmusculus.UCSC.mm10.knownGene
> promoter <- getPromoters(TxDb=txdb, upstream=3000, downstream=3000)
> tagMatrixList <- lapply(peaks, getTagMatrix, windows=promoter)
> plotAvgProf(tagMatrixList, xlim=c(-3000, 3000))
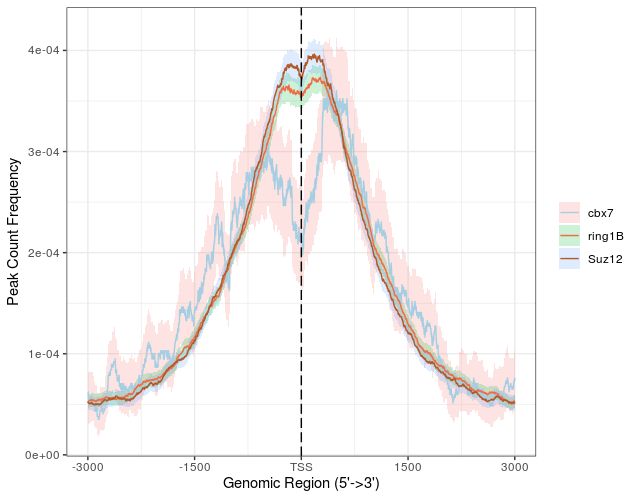
还支持使用bootstrap估计置信区间:
plotAvgProf(tagMatrixList, xlim=c(-3000, 3000), conf = 0.95, resample = 1000)
注释peak
ChIPseeker负责注释的就是annotatePeak
#对于非向量的循环,推荐构建list,然后用lapply
> peaks <- list(ring1B, cbx7, Suz12)
#注释。启动子区域是没有明确的定义的,所以你可能需要指定tssRegion,把基因起始转录位点的上下游区域来做为启动子区域。
> peakAnnoList <- lapply(peaks, annotatePeak, tssRegion=c(-2500,2500), TxDb=TxDb.Mmusculus.UCSC.mm10.knownGene,
addFlankGeneInfo=TRUE, flankDistance=5000)
#结果可以用as.GRanges或者as.data.frame查看
#Granges是R语言中的数据结构,它主要用来存储基因组中的坐标区间。
> as.GRanges(peakAnnoList[[1]])
> as.data.frame(peakAnnoList[[1]])
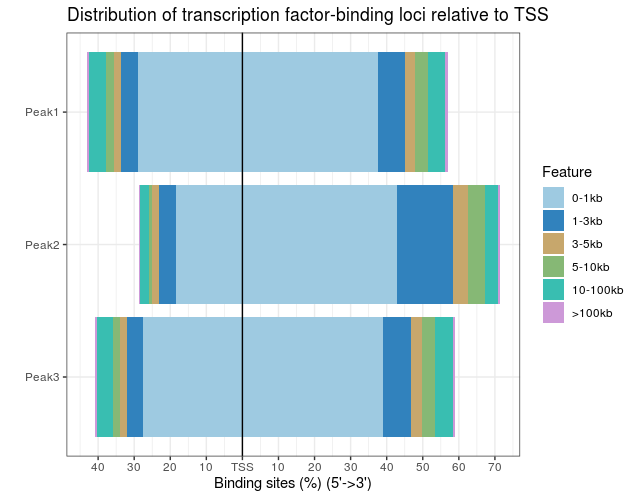
#画图,不同蛋白的靶位点的注释情况
> plotDistToTSS(peakAnnoList)
韦恩图
genes= lapply(peakAnnoList, function(i) as.data.frame(i)$geneId)
vennplot(genes[1:2:])

注释的可视化:
#先注释
peakAnno <- annotatePeak(cbx7, tssRegion=c(-3000, 3000),
TxDb=txdb, annoDb="org.Mm.eg.db")
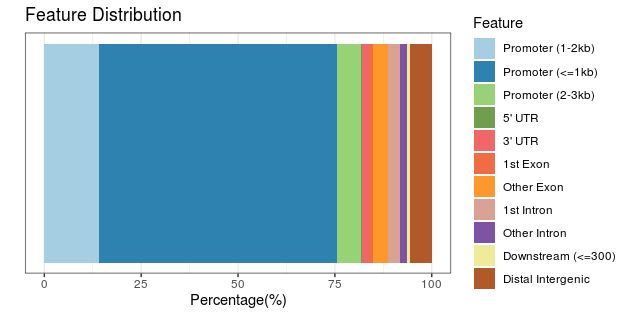
#柱形图
plotAnnoBar(peakAnno)
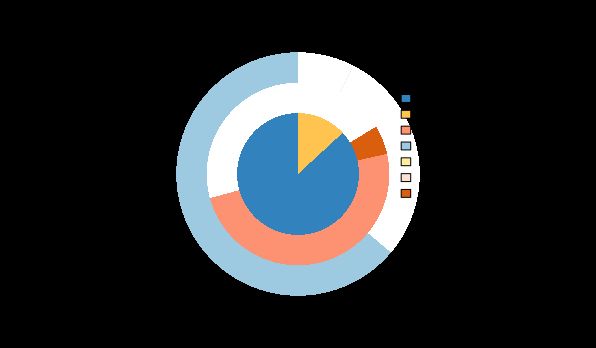
#vennpie
vennpie(peakAnno)
#饼图
plotAnnoPie(peakAnno)

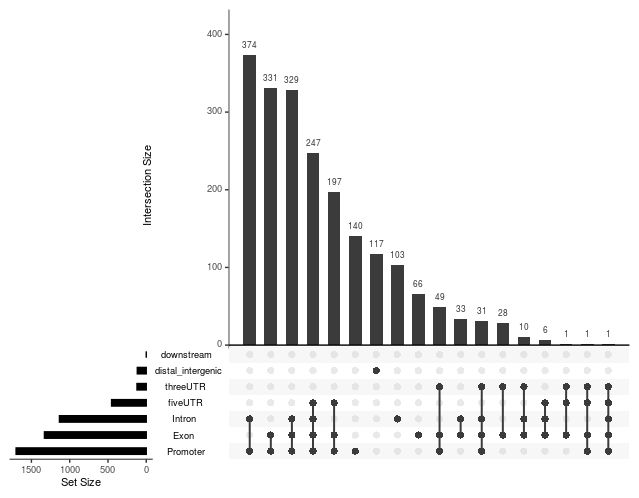
# 展示注释信息的交集
$ upsetplot(peakAnno)
7.deeptools的使用
BAM文件是SAM的二进制转换版,应该都知道。那么bigWig格式是什么?bigWig是wig或bedGraph的二进制版,存放区间的坐标轴信息和相关计分(score),主要用于在基因组浏览器上查看数据的连续密度图,可用wigToBigWig从wiggle进行转换。为什么要用bigWig? 主要是因为BAM文件比较大,直接用于展示时对服务器要求较大。因此在GEO上仅会提供bw,即bigWig下载,便于下载和查看。
(一)将bam文件转换成bw(bigwig)文件
这一步需要用到的是deeptool里的bamCoverage工具:
#!/bin/bash
bamCoverage -b cbx7.bam -o cbx7.bw -p 10 --normalizeUsing RPKM
bamCoverage -b ring1B.bam -o ring1B.bw -p 10 --normalizeUsing RPKM
bamCoverage -b Suz12.bam -o Suz12.bw -p 10 --normalizeUsing RPKM
bamCoverage -b IgGold.bam -o IgGold.bw -p 10 --normalizeUsing RPKM
-b:指要进行操作的bam文件,后面跟的是bam文件名。
-o:指输出的文件,后面跟的是要输出的文件名。
-p: --numberOfProcessors,进程的数量。
--normalizeUsing: 标准化的方法。这个工具有RPKM, CPM, BPM, RPGC, None几个不同的选择,这里我选了RPKM。
(二)peak分布可视化
也是deeptool里的工具。为了统计全基因组范围的peak在基因特征的分布情况,需要用到computeMatrix计算,用plotHeatmap以热图的方式对覆盖进行可视化,用plotProfile以折线图的方式展示覆盖情况。
computeMatrix有两种模式,scale-regions mode和reference-point mode(参考文章:https://www.jianshu.com/p/2b386dd437d3)
reference-point(relative to a point): 计算某个点的信号丰度。给定一个bed file,以某个点为中心开始统计信号(TSS/TES/center)。
scale-regions(over a set of regions): 把所有基因组区段缩放至同样大小,然后计算其信号丰度。简单来说会将给定bed file范围内的结合信号做一个统计(指的是一段长度),并将基因长度统一scale到设定regionBdoyLength的长度,加上统计基因上游和下游Xbp的信号(beforeRegionStartLength参数和afterRegionStartLength参数)
不管是哪一种,有两个参数是必须的,-S提供bigwig文件,-R提供基因的注释信息。
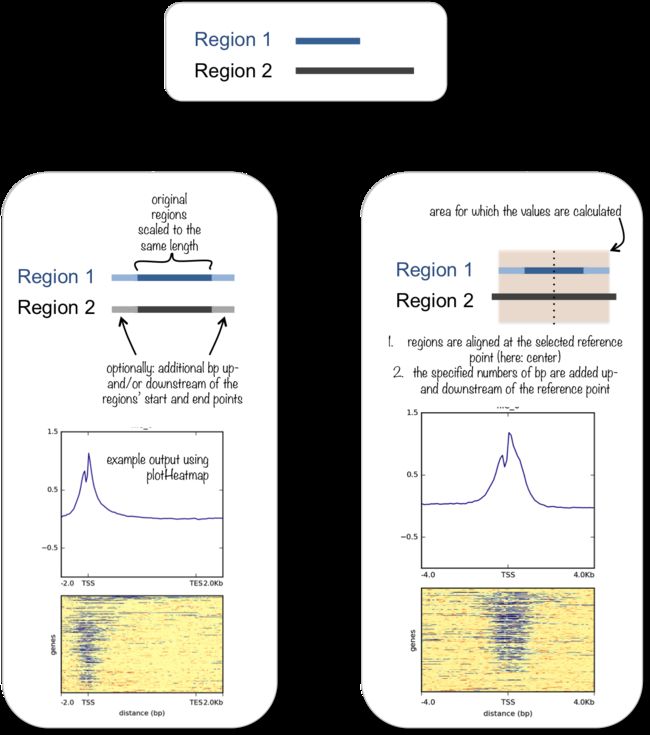
如果想得到单独的文件,就是说每个样品都有单独的matrix文件,可以用下面的代码:
#!/bin/bash
computeMatrix reference-point --referencePoint TSS -a 2500 -b 2500 -S /media/yanfang/FYWD/CHIPSEQ/bw/cbx7.bw -R /media/yanfang/FYWD/CHIPSEQ/bw/mm10.bed --skipZeros -o cbx7_matrix_TSS.gz
computeMatrix reference-point --referencePoint TSS -a 2500 -b 2500 -S /media/yanfang/FYWD/CHIPSEQ/bw/ring1B.bw -R /media/yanfang/FYWD/CHIPSEQ/bw/mm10.bed --skipZeros -o ring1B_matrix_TSS.gz
computeMatrix reference-point --referencePoint TSS -a 2500 -b 2500 -S /media/yanfang/FYWD/CHIPSEQ/bw/Suz12.bw -R /media/yanfang/FYWD/CHIPSEQ/bw/mm10.bed --skipZeros -o Suz12_matrix_TSS.gz
computeMatrix reference-point --referencePoint TSS -a 2500 -b 2500 -S /media/yanfang/FYWD/CHIPSEQ/bw/IgGold.bw -R /media/yanfang/FYWD/CHIPSEQ/bw/mm10.bed --skipZeros -o IgGold_matrix_TSS.gz
reference-point: 选择模式,这里我选择了第二种。
-p 10:指10个进程。我的电脑比较渣,所以没有加这个参数。
--referencePoint TSS:选择参考点,这里选择转录起始位点。
-a 3000:下游3000bp
-b 3000:上游3000bp
-S:指bw文件,后面跟的是文件的位置。
-R:参考基因组的注释文件,这里要求是bed格式的。后面跟的是文件的位置。
--skipZeros:Whether regions with only scores of zero should be included or not. Default is to include them.
-o:输出文件,后面跟的是输出文件的名字
如果想把四个样品合在一起,后续画图也是merge在一起的,可以用下面的代码:
$ computeMatrix reference-point --referencePoint TSS -a 2500 -b 2500 -S /media/yanfang/FYWD/CHIPSEQ/bw/*.bw -R /media/yanfang/FYWD/CHIPSEQ/bw/mm10.bed --skipZeros -o merge_matrix_TSS.gz
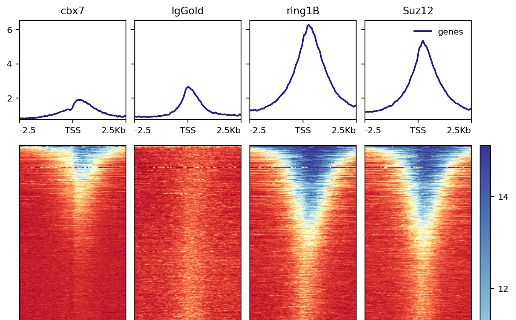
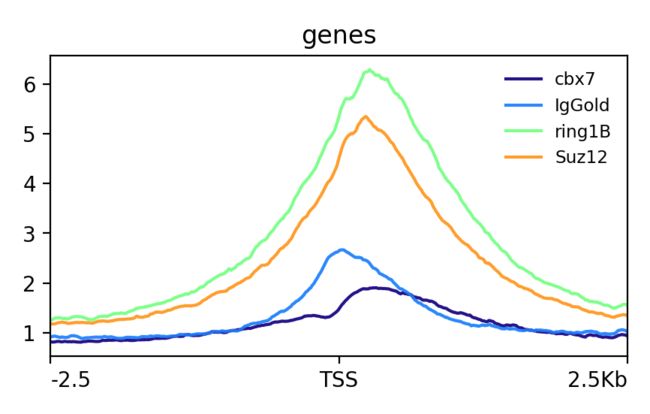
画基因的TSS附近的profile和heatmap图
#heatmap图
$ plotHeatmap -m merge_matrix_TSS.gz -out merge_heatmap.png
#将heatmap图输出为pdf格式
$ plotHeatmap --dpi 720 -m merge_matrix_TSS.gz -out merge_heatmap.pdf --plotFileFormat pdf
#轮廓图
plotProfile -m merge_matrix_TSS.gz -out merge_profile.png
#将轮廓图输出为pdf格式
$ plotProfile --dpi 720 -m merge_matrix_TSS.gz -out merge_profile.pdf --plotFileFormat pdf
#如果想把每个样品的轮廓图merge上在一个图片里,在代码后面加上--perGroup
$ plotProfile -m merge_matrix_TSS.gz -out merge_profile.png --perGroup
$ plotProfile --dpi 720 -m merge_matrix_TSS.gz -out merge_profile.pdf --plotFileFormat pdf --perGroup
也可以整合所有的chipseq的bw文件,画基因的genebody附近的profile和heatmap图(图就不放上来了):
$ computeMatrix scale-regions -a 3000 -b 3000 -m 5000 -S /media/yanfang/FYWD/CHIPSEQ/bw/*.bw -R /media/yanfang/FYWD/CHIPSEQ/bw/mm10.bed --skipZeros -o merge_scaleregion_matrix_TSS.gz
$ plotHeatmap -m merge_scaleregion_matrix_TSS.gz -out merge_scaleregion_matrix_TSS.png
$ plotProfile -m merge_scaleregion_profile_TSS.gz -out merge_scaleregion_profile_TSS.png