第十章
使用序列数据
生物信息学的核心问题之一是处理大量的(通常定义糟糕或模糊)文件格式。久而久之,一些特定的简单的人类可读的格式成为了事实上的标准。
- 彼得·科克等, 2010年
好的程序员知道怎样写程序。杰出的程序员知道怎样重写(并重新使用)。
- 大教堂和集市 埃里克·斯蒂芬·雷蒙德
在生物信息学中,核苷酸(和蛋白质)序列普遍以两种纯文本格式存储:FASTA和FASTQ,发音分别为fast-ah(或fast-A)和fast-Q。 我们将在本节讨论这两种格式和它们各自的局限性,然后了解一些处理这些数据格式的工具。这是一个简短的章节,但是本节有一个重要的事项需要注意,也就是处理这些特定生物信息学格式的共同误区。即使像文件格式这样的细枝末节中存在的简单错误,也需要很多时间和精力去发现和修复,所以请尽早记住这些细节。
FASTA格式
FASTA格式源于由William R. Pearson和David J. Lipman开发的FASTA比对套装软件。FASTA格式用于存储各种序列数据,不需要每个碱基对的质量分数。这些序列数据包括参考基因组文件,蛋白质序列,DNA编码序列(CDS),转录本序列等。FASTA也可以用来存储多序列比对数据,但是我们这里不会讨论这个FASTA的格式的特殊变体。我们在前面的章节遇到了FASTA格式,但在本节中,我们将详细介绍这种格式,了解该格式常见的缺陷,并介绍一些处理这种格式的工具。
FASTA文件由序列条目组成,每个条目都包含两部分内容:序列描述和序列数据。描述行以一个大于号(>)开始,包含序列标识符和其他(可选)信息。 序列数据从描述行的下一行开始,直到另一个描述行(以 > 开头的行)出现或文件结束。 本章GitHub目录中的 egfr_ FL ank.fasta 文件是一个FASTA例子文件:
$ head -10 egfr_flank.fasta
> ENSMUSG00000020122 | ENSMUST00000138518
CCCTCCTATCATGCTGTCAGTGTATCTCTAAATAGCACTCTCAACCCCCGTGAACTTGGT
TATTAAAAACATGCCCAAAGTCTGGGAGCCAGGGCTGCAGGGAAATACCACAGCCTCAGT
TCATCAAAACAGTTCATTGCCCAAAATGTTCTCAGCTGCAGCTTTCATGAGGTAACTCCA
GGGCCCACCTGTTCTCTGGT
> ENSMUSG00000020122 | ENSMUST00000125984
GAGTCAGGTTGAAGCTGCCCTGAACACTACAGAGAAGAGAGGCCTTGGTGTCCTGTTGTC
TCCAGAACCCCAATATGTCTTGTGAAGGGCACACAACCCCTCAAAGGGGTGTCACTTCTT
CTGATCACTTTTGTTACTGTTTACTAACTGATCCTATGAATCACTGTGTCTTCTCAGAGG
CCGTGAACCACGTCTGCAAT
FASTA格式的简单性和灵活性却带来了令人遗憾的缺点:FASTA格式是一个松散定义的特殊格式(虽然这种现象在生物信息学中十分常见)。因此,你可能会遇到FASTA格式的各种变体,这些变体会造成微小的错误,除非你的程序足够强大。这就是为什么最好使用现有的FASTA/FASTQ解析库而不是执行自定义的解析库;现有的库已经过开源社区审查(后续将更多讨论这一部分内容)。
关于FASTA格式最令人头疼的是描述行中标识符的格式没有通用的规范。例如,如下FASTA描述行是否指代同一条目?
>ENSMUSG00000020122|ENSMUST00000138518
> ENSMUSG00000020122|ENSMUST00000125984
>ENSMUSG00000020122|ENSMUST00000125984|epidermal growth factor receptor
>ENSMUSG00000020122|ENSMUST00000125984|Egfr
>ENSMUSG00000020122|ENSMUST00000125984|11|ENSFM00410000138465
没有标识符的标准方案,我们不能使用简单的精确匹配确定一个标识符是否和FASTA条目的标题行匹配。相反,我们需要依靠FASTA文件描述行和标识符之间的模糊匹配。这可能会使得判断标准变得相当凌乱:匹配模式的宽容度应该是多少?如果正则表达式过于宽泛是否会匹配到错误的序列?基本上来说,模糊匹配是一个脆弱的策略。
幸运的是,这个问题有更好的解决方案(也很简单):与其事后依靠模糊匹配来纠正不一致的命名,还不如一开始就拥有严格的命名规范并始终保持一致。然后,在运行任何外部来源的数据时,需要通过几个合理性检查,以确保数据遵循对应的格式。 这些检查不需要太复杂(检查重复的名称,手动检查一些条目,检查 > 和标识符之间错误的空格,检查不同文件之间名字的重合等)。
如果你需要整理外部数据,请始终保留原始文件并编写脚本将更正的版本写入新的文件。这样,脚本可以轻松地重新运行,用于处理你获得的新版本的原始数据集(但你仍然需要检查每一项内容-不要盲目信任数据!)。
常见的命名规范是将描述行利用第一个空格分为两部分:标识符和注释。此格式的序列通常如下所示:
> gene_00284728 length = 231; type = dna
GAGAACTGATTCTGTTACCGCAGGGCATTCGGATGTGCTAAGGTAGTAATCCATTATAAGTAACATGCGCGGAATATCCG
GAGGTCATAGTCGTAATGCATAATTATTCCCTCCCTCAGAAGGACTCCCTTGCGAGACGCCAATACCAAAGACTTTCGTA
GCTGGAACGATTGGACGGCCCAACCGGGGGGAGTCGGCTATACGTCTGATTGCTACGCCTGGACTTCTCTT
这里 gene_00284728 是标识符,length = 231; type = dna 是注释。此外,ID应该是唯一的。尽管不是标准规范,但是将第一个空格之前的内容作为标识符,之后的内容作为非必要注释在生物信息学程序中是很常见的(例如,BEDtools,Samtools和BWA都是这样处理的)。有了这个规范以后,通过标识符查找一个特定的序列会很容易,我们将在本章末尾讨论如何高效地利用这种方式处理经过索引的FASTA文件。
FASTQ格式
FASTQ格式通过给序列中每个碱基添加数字质量分数的形式扩展了FASTA文件。FASTQ格式被广泛用于存储高通量测序数据,该数据利用每个碱基的质量分数来表示每个碱基测序的可信度。然而和FASTA格式一样,FASTQ格式也拥有变体和缺陷,会使看似简单的格式变得很难处理。
FASTQ格式如下所示:
@ DJB775P1:248:D0MDGACXX:7:1202:12362:49613 ①
TGCTTACTCTGCGTTGATACCACTGCTTAGATCGGAAGAGCACACGTCTGAA ②
+ ③
JJJJJIIJJJJJJHIHHHGHFFFFFFCEEEEEDBD?DDDDDDBDDDABDDCA ④
@ DJB775P1:248:D0MDGACXX:7:1202:12782:49716
CTCTGCGTTGATACCACTGCTTACTCTGCGTTGATACCACTGCTTAGATCGG
+
IIIIIIIIIIIIIIIHHHHHHFFFFFFEECCCCBCECCCCCCCCCCCCCCCC
① 描述行,以@符号开始。包含标识符记录和其他信息。
② 序列数据,可以是一行或多行。
③ 以 + 开头的行,位于序列数据之后,表示序列的结束。在旧版本的FASTQ文件中,通常会这里重复描述行,但这是多余的,会导致不必要的FASTQ文件过大。
④ 质量数据,也可以是一行或多行,但必须和序列长度相同。每个碱基质量数值以ASCII字符编码,编码规则我们将在后文讨论(“碱基质量”见344页)。
与FASTA文件一样,FASTQ文件中一个常见的规范是将描述行利用第一个空格分解为两部分:标识符记录和注释。
如何正确地解析FASTQ文件其实是非常棘手的。一个常见的误区是将每一个以 @ 符号开始的行都当作说明行。然而,@ 本身也是一个有效的质量符号。FASTQ的序列行和质量行可以延续到下一行,所以以 @ 符号开头的行可能是质量行,而不是标题行。所以,写一个始终把以 @ 开头的行作为标题行的解析程序会导致不正确的解析。但是,我们可以利用质量分数的字符数必须等于序列的字符数这一准则来准确解析这种格式,这其实就是后文中 readfq 解析器的工作原理。
计算FASTA/FASTQ条目的输入和输出
作为纯文本格式,可以轻松使用Unix工具处理FASTQ和FASTA文件。一个常见的生物信息学命令行是这样的:
$ grep -c "^>" egfr_flank.fasta
5
正如本书47页“Pipes实战:利用grep和pipes创建简单的程序”所说,你必须引用** > **字符来防止shell把它当作一个重定向运算符(并覆盖你的FASTA文件!)。这是一个保险的计算FASTA文件数目的方式,因为虽然格式定义宽松,但是每个序列都具有一个描述行,只有这些描述行以 > 符号开始 。
我们可能会尝试使用类似的方法处理FASTQ文件,用 @ 符号代替 >:
$ grep -c "^@" untreated1_chr4.fq
208779
这意味着 untreated1_chr4.fq 有208779个条目。但是,仔细检查 untreated1_chr4.fq 文件,你会发现每个FASTQ条目占用四行,但总行数是:
$ wc -l untreated1_chr4.fq
817420 untreated1_chr4.fq
而817420/4 = 204355,这和 grep -c 命令得到的结果有很大不同!这是发生了什么呢?请记住,@ 是一个有效的质量符号,质量行可以以这个符号开始,可以使用 grep "^@" untreated1_chr4.fq | less 命令行来查看这样的例子。
如果你很确定你的FASTQ文件每个序列条目占用四行,可通过 wc -l 命令估计文件行数并除以4的方式来估计序列条目。如果你不确定某些FASTQ条目是否包含多行,一个更强大的计算序列条目的方法是bioawk:
$ bioawk -cfastx 'END {print NR}' untreated1_chr4.fq
204355
核苷酸编码
随着基础FASTA / FASTQ格式的覆盖,让我们来看看以这些格式出现的核苷酸和碱基质量分数的标准编码。 显然,编码核苷酸很简单:A,T,C,G分别代表核苷酸腺嘌呤,胸腺嘧啶,胞嘧啶和鸟嘌呤。小写的碱基经常被用来代表“软覆盖”重复序列或低复杂度序列(RepeatMasker 和 Tandem Repeats Finder 程序均采用这种格式)。重复序列和低复杂度的序列也可以被“硬覆盖”,核苷酸序列被替换为N(有时是X)。
简并(或模糊)核苷酸编码被用于表示两个或多个碱基。例如,N可以用于表示任何碱基。国际纯粹与应用化学联合会(IUPAC)拥有一组标准化的核苷酸编码,既包括明确的也包括简并的(见表10-1)。
表 10-1 IUPAC 核苷酸代码
| IUPAC代码 | 碱基 | 助记符 |
|---|---|---|
| A | Adenine | 腺嘌呤 |
| T | Thymine | 胸腺嘧啶 |
| C | Cytosine | 胞嘧啶 |
| G | Guanine | 鸟嘌呤 |
| N | A, T, C, G | 任何碱基 |
| Y | C, T | 嘧啶 |
| R | A, G | 嘌呤 |
| S | G, C | 强键 |
| W | A, T | 弱键 |
| K | G, T | 酮基 |
| M | A, C | 氨基 |
| B | C, G, T | A以外的所有碱基,位于A碱基后 |
| D | A, G, T | C以外的所有碱基,位于C碱基后 |
| H | A, C, T | G以外的所有碱基,位于G碱基后 |
| V | A, C, G | T或U(尿嘧啶)以外的所有碱基,位于U碱基后 |
有些生物信息学程序对于简并核苷酸的处理可能会有所不同。例如,BWA读长比对工具可以将参考基因组中的简并核苷酸字符转换为随机碱基(Li and Durbin, 2009),但是由于使用了随机种子集合,所以不会在重新索引比对数据的时候产生两种不同的结果。
碱基质量
FASTQ条目的每个序列碱基在质量行都具有相应的数字质量分数。每个碱基的质量分数都编码为一个ASCII字符。质量行看起来就像一串随机字符,如第四行所示:
@ AZ1:233:B390NACCC:2:1203:7689:2153
GTTGTTCTTGATGAGCCATGAGGAAGGCATGCCAAATTAAAATACTGGTGCGAATTTAAT
+
CCFFFFHHHHHJJJJJEIFJIJIJJJIJIJJJJCDGHIIIGIGIJIJIIIIJIJJIJIIH
(这个FASTQ条目在本章的 README 文件中,如果你想按照这里所说的跟着一起看看。)
记住,ASCII字符仅在内部表示为0到127之间的整数(详情参见 man ascii 命令)。因为并不是所有的ASCII字符都可以在屏幕上显示出来(例如,字符呼应 "\ 07" 会发出“叮”的声音),质量分数仅限于可打印的ASCII字符,范围从33到126(代表空格的字符32被省略)。
所有编程语言都具有将字符转换为十进制ASCII码和将十进制ASCII码转换为字符的功能。在Python中,分别对应函数 ord() 和 chr()。让我们使用 ord() 函数在Python交互式解释器中将质量字符转换为十进制ASCII码列表:
>>> qual = "JJJJJJJJJJJJGJJJJJIIJJJJJIGJJJJJIJJJJJJJIJIJJJJHHHHHFFFDFCCC"
>>> [ord(b) for b in qual]
[74, 74, 74, 74, 74, 74, 74, 74, 74, 74, 74, 74, 71, 74, 74, 74, 74, 74, 73,
73, 74, 74, 74, 74, 74, 73, 71, 74, 74, 74, 74, 74, 73, 74, 74, 74, 74, 74,
74, 74, 73, 74, 73, 74, 74, 74, 74, 72, 72, 72,
67, 67, 67]
遗憾的是,将这些ASCII数值转换成有意义的质量分数可能非常棘手,因为目前存在三种不同的质量体系:Sanger,Solexa和Illumina(见表10-2)。负责Biopython,BioPerl和BioRuby这类项目的开放性生物信息学基金会(OBF),将它们命名为 fastq-sanger,fastq-solexa 和 fastaq-illumina。值得庆幸的是,生物信息学领域最终似乎确定统一使用Sanger编码(也就是这里显示的质量行格式),因此我们将使用此方案逐步完成转换过程。
表 10-2 FASTQ质量体系(来源 Cock et al., 2010,已获得许可)
| 名称 | ASCII字符范围 | 偏移 | 质量分数类型 | 质量分数范围 |
|---|---|---|---|---|
| Sanger, Illumina(版本1.8以上) | 33-126 | 33 | PHRED | 0-93 |
| Solexa, 早期Illumina(版本1.3之前) | 59-126 | 64 | Solexa | 5-62 |
| Illumina(版本1.3-1.7) | 64-126 | 64 | PHRED | 0-62 |
首先,我们需要减去一个偏移值来将这个Sanger质量分数转换为PHRED质量分数。PHRED是由Phil Green撰写的早期碱基调用程序,用于解析他自己写的荧光追踪数据。通过查看表10-2,我们注意到Sanger格式的偏移量为33,因此我们从每个质量分数中减去33:
>>> phred = [ord(b)-33 for b in qual]
>>> phred
[41, 41, 41, 41, 41, 41, 41, 41, 41, 41, 41, 41, 38, 41, 41, 41, 41, 41, 40,
40, 41, 41, 41, 41, 41, 40, 38, 41, 41, 41, 41, 41, 40, 41, 41, 41, 41, 41,
41, 41, 40, 41, 40, 41, 41, 41, 41, 39, 39, 39, 39, 39, 37, 37, 37, 35, 37,
34, 34, 34]
现在,鉴于我们的Sanger质量分数已经转换为PHRED质量分数,我们就可以利用下面的公式将质量分数转换为碱基测序正确的估计概率:
将概率转化为质量,我们使用这个函数的倒数:
在我们的例子中,我们需要前一个方程式。将其应用于我们的PHRED质量分数:
>>> [10**(-q/10) for q in phred]
[1e-05, 1e-05, 1e-05, 1e-05, 1e-05, 1e-05, 1e-05, 1e-05, 1e-05, 1e-05, 1e-05,
1e-05, 0.0001, 1e-05, 1e-05, 1e-05, 1e-05, 1e-05, 0.0001, 0.0001, 1e-05,
1e-05, 1e-05, 1e-05, 1e-05, 0.0001, 0.0001, 1e-05, 1e-05, 1e-05, 1e-05,
1e-05, 0.0001, 1e-05, 1e-05, 1e-05, 1e-05, 1e-05, 1e-05, 1e-05, 0.0001,
1e-05, 0.0001, 1e-05, 1e-05, 1e-05, 1e-05, 0.0001, 0.0001, 0.0001, 0.0001,
0.0001, 0.0001, 0.0001, 0.0001, 0.0001, 0.0001, 0.0001, 0.0001, 0.0001]
公式。关于这个格式的更多详情,请参阅Cock et al., 2010。
示例:检查和去除低质量碱基
请注意前一个例子中碱基的准确度是如何下降的;这是Illumina测序的特征误差分布。本质上来说,我们通过这种测序技术获得的读长的碱基测序错误概率逐渐增加(朝向3'端)。这可能对下游分析产生深远的影响!当处理测序数据时,你应该总是
- 了解测序技术的错误分布和局限性(例如,是否受到GC含量的影响)
- 考虑上述内容将如何影响你的分析
所有这一切都是实验特异性的,而且需要精心的规划。
把我们关于碱基准确度的Python列表作为一个学习工具是很用的,可以看到如何将概率转换为质量,但它不能帮助我们了解数百万序列的质量概况。从这个意义上来说,一图抵千字-有软件可以帮助我们看到读长中碱基的质量分布。最流行的是Java程序 FastQC,这款软件很容易运行并输出有用的图形和质量指标。如果你喜欢使用R语言,你可以使用Bioconductor中的软件包 qrqc(由鄙人所写)。我们将在示例中使用 qrqc 软件包,这样我们就可以自己了解如何可视化这些数据。
我们先来安装运行这个例子所有必要的程序。首先,利用如下命令行在R中安装 qrqc 软件包:
> library(BiocInstaller)
> biocLite('qrqc')
接下来,让我们来安装两个程序用来去除低质量碱基:sickle 和 seqtk 。seqtk 是由Heng Li编写的一个通用序列工具包 - 拥有在序列末端去除低质量碱基的子命令(还有许多有用的其他功能)。利用Homebrew软件包管理系统,sickle 和 seqtk 很容易在Mac OS X操作系统上安装(例如,利用brew install seqtk 和 brew install sickle 命令行)。
在安装好这些程序之后,让我们来开始修整在GitHub存储库中本章目录下的 untreated1_chr4.fq FASTQ文件。这个FASTQ文件由Bioconductor中 pasillaBamSubset 软件包的 untreated1_chr4.bam BAM文件生成(更多信息请参见本章目录中的 README 文件)。为了保持事情简单,我们将使用每个程序的默认设置。从 sickle 软件包开始:
$ sickle se -f untreated1_chr4.fq -t sanger -o untreated1_chr4_sickle.fq
FastQ records kept: 202866
FastQ records discarded: 1489
sickle 通过 -f 指定输入文件,通过 -t 指定质量分数类型,通过 -o 得到修整后的文件。
现在,让我们来运行 seqtk trimfq 命令,这个命令需要一个参数并以标准形式输出修整后的序列:
$ seqtk trimfq untreated1_chr4.fq > untreated1_chr4_trimfq.fq
让我们在R中比较上述结果。我们使用 qrqc 软件包根据位置收集这些文件中的碱基质量分布,然后使用 ggplot2 软件包可视化这些结果。我们可以逐一加载这些软件包,但一个不错的工作流程是利用 lapply() 函数自动执行:
# trim_qual.R -- 检查修剪前后碱基质量
library(qrqc)
# FASTQ文件
fqfiles <- c(none="untreated1_chr4.fq",
sickle="untreated1_chr4_sickle.fq",
trimfq="untreated1_chr4_trimfq.fq")
# 使用 qrqc 软件包的 readSeqFile 函数加载每个文件
# 我们只需要碱基质量,所以关掉
# readSeqFile 函数的一些其他功能。
seq_info <- lapply(fqfiles, function(file) {
readSeqFile(file, hash=FALSE, kmer=FALSE)
})
# 将碱基质量提取为数据框,并追加
# 一列使用修剪工具(或没有使用)的情况。这部分
# 在后面的作图中使用。
quals <- mapply(function(sfq, name) {
qs <- getQual(sfq)
qs$trimmer <- name
qs
}, seq_info, names(fqfiles), SIMPLIFY=FALSE)
# 将列表中单独的数据框合并为单个数据框
d <- do.call(rbind, quals)
# 碱基质量可视化
p1 <- ggplot(d) + geom_line(aes(x=position, y=mean, linetype=trimmer))
p1 <- p1 + ylab("mean quality (sanger)") + theme_bw()
print(p1)
# 使用 qrqc 软件包的 qualPlot 函数与列表生成面板图
# 只显示 10% 到 90% 的分位数和低通曲线
p2 <- qualPlot(seq_info, quartile.color=NULL, mean.color=NULL) + theme_bw()
p2 <- p2 + scale_y_continuous("quality (sanger)")
print(p2)
该脚本会生成两幅图:图10-1和图10-2。在图10-2中我们可以同时看到两个修剪软件对数据碱基质量分布的影响:通过去除低质量碱基,我们进一步缩小了读长碱基的质量分布范围。在图10-1中,我们可以发现去除低质量碱基增加了整个读长的平均质量,但仍然可以看到碱基质量沿着读长末端逐渐下降。
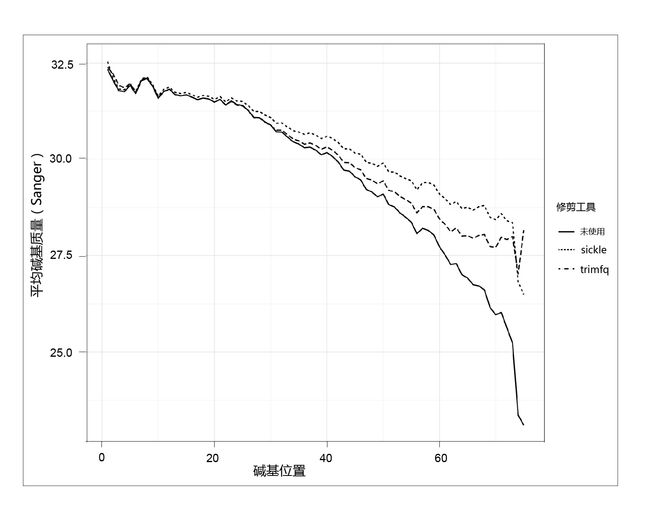
图 10-1 修剪前以及利用 sickle 和 seqtk trimfq 处理后的读长平均碱基质量

图 10-2 修剪前以及利用 sickle 和 seqtk trimfq 处理后每个碱基位置的 10% 到 90% 分位数和低通曲线
在一行序列中,我们可以从末尾去除低质量的碱基-修剪命令并不困难。更重要的步骤是通过比较去除低质量碱基之前和之后的文件来可视化这些修剪程序对我们的数据做了怎样的修改。检查程序如何更改我们的数据而不是盲目相信它们才是强大的生物信息学的重要组成部分,也是遵守黄金法则(不要信任你的工具)的表现。在这个例子中,检查一小部分数据只需要不超过20行代码(忽略空白行和提高可读性的注释)和几分钟的时间,但关于这些程序对我们的数据做了什么和程序之间的差异,这些步骤给了我们有价值的理解。如果我们愿意,我们也可以更改不同的设置来同时运行这两个质量修饰程序,并比较这些设置对结果的影响。大部分严谨的生物信息学都是这样的过程:运行程序,对比原始数据与输出结果,运行程序,比较输出结果,如此往复。
FASTA/FASTQ解析示例:核苷酸计数
编写你自己的FASTA/FASTQ解析器其实并不是很困难,事实上,这是一个非常有教育性的编程练习。 但是当我们需要使用解析器来处理真实的数据时,最好还是使用可靠的现有库。记住本章开头引用的话:杰出的程序员知道何时重用代码。有很多开源的,免费提供的,可靠的解析器已经经过了开源社区审核,并且旨在加速解析过程。我们将使用Heng Li的 readfq 应用程序,因为它可以解析FASTA和FASTQ文件,使用起来很简单,是独立应用(意味着它不需要安装任何依赖项)。Biopython和BioPerl这两个流行的程序库中有很好的可选FASTA/FASTQ解析器。
我们将使用Python版本的 readfq 程序,readfq.py。你可以从GitHub上下载这个Python文件(本书的GitHub资源库中也有这个文件的副本)。虽然我们可以使用 from readfq import readfq 命令来执行 readfq.py 中的FASTA/FASTQ解析程序,但是对于单文件脚本其实更简单,你可以直接将程序复制粘贴到你的脚本中。在本书中,我们使用 from readfq import readfq 命令来避免占据例子文件的空间。
readfq 程序的 readfq() 函数使用简单。readfq() 采用一个文件对象(例如,一个已经通过
open('filename.fa') 或 sys.stdin 打开的文件名称),并会产生FASTQ/FASTA条目,直到它到达文件或流的末尾。每个FASTQ/FASTA条目由 readfq() 函数返回为包含条目描述,序列和质量的元组。如果 readfq() 正在读取一个FASTA文件,质量行是无。这就是利用 readfq() 函数读取FASTQ/FASTA序列行的所有内容。

我们编写一个简单的程序来计算文件中每个IUPAC核苷酸的数量:
#!/usr/bin/env python
# nuccount.py -- 一个文件中的所有核苷酸数目
import sys
from collections import Counter ①
from readfq import readfq
IUPAC_BASES = "ACGTRYSWKMBDHVN-." ②
# 初始化计数器
counts = Counter() ③
for name, seq, qual in readfq(sys.stdin): ④
# 对于每一个序列条目,向计数器添加所有的碱基数量
counts.update(seq.upper()) ⑤
#打印结果
for base in IUPAC_BASES:
print base + "\t" + str(counts[base]) ⑥
① 我们使用 collections 模块中的 Counter 计数核苷酸。Counter 的工作原理很像Python的 dict 类(技术上来说,它是 dict 的一个子类),具有使计数更容易的附加功能。
② 这个全局变量定义了所有的IUPAC核苷酸,我们稍后将用它来打印顺序一致的碱基(因为像Python字典一样,Counter 对象不保持顺序)。在脚本的顶部放置类似IUPAC_BASES的大写常量让读者清楚是一个很好的做法。
③ 创建一个新的 Counter 对象。
④ 此行使用 readfq 模块中的 readfq 函数从文件句柄参数读取FASTQ/FASTQ条目(在这种情况下,sys.stdin,标准输入)转换成 name,seq 和 qual 变量(通过Python中被称为元组拆封的功能)。
⑤ Counter.update() 方法读取任何可迭代的对象(在此情况下,序列碱基的字符串),并将它们添加到计数器中。我们也可以使用for循环遍历 seq 变量的每个字符,利用 counts[seq.upper()] 命令递增计数。请注意,我们用 upper() 函数把所有的字符转换为大写,因此小写的“软覆盖”碱基也会被计数。
⑥ 最后,我们迭代所有的IUPAC碱基并输出它们的数量。
这个版本需要通过标准输入获得输入文件,所以保存此文件并使用 chmod + X nuccount.py 命令添加执行权限后,我们可以通过下述命令运行:
$ cat contam.fastq | ./nuccount.py
A 103
C 110
G 94
T 109
R 0
Y 0
S 0
W 0
K 0
M 0
B 0
D 0
H 0
V 0
N 0
- 0
. 0
请注意,我们不一定要让Python脚本可执行;另一个选择是简单地用 python nuccount.py 命令启动它 。 无论哪种方式都会得到相同的结果,所以这最终是一种风格的选择。但是,如果你确实想让它变成一个可执行的脚本,记住要做以下的事情:
- 包含标识行
#!/usr/bin/env python - 使用
chmod + X命令使脚本可执行
这个脚本有很多可以改进的地方:添加对每个序列碱基组成统计的支持,采用文件参数而不是通过标准输入,“软覆盖”碱基(小写)计数,CpG位点计数或在文件中发现非IUPAC核苷酸时发出警告。核苷酸计数很简单-脚本中最复杂的部分是 readfq() 函数。这是重用代码之美:书写良好的函数和库让我们不需要重写复杂的解析器。相反,我们使用更广泛的社区合作开发的软件,测试,排除故障,使其更有效率(参见 readfq 软件在GitHub上的提交历史作为例子)。重用软件不是欺骗-这是行家写程序的方式。
索引FASTA文件
我们经常需要对FASTA文件的子序列进行有效的随机访问(给定区域)。乍一看来,写一个脚本来完成似乎并不困难。例如,我们可以编写一个遍历FASTA条目的脚本,提取与指定区域重叠的序列。但是,在提取一些随机序列时,这不是一个有效的方法。为了知道为什么,假设我们需要访问小鼠基因组第8号染色体(123,407,082至123,410,742区域的)序列。这种方法会不必要地在内存中解析和加载1号染色体至7号染色体的数据,即使我们不需要从这些染色体中提取子序列。从磁盘中读取全部染色体并将其复制到内存中可能效率相当低下-我们为了提取3.6 kb数据必须加载所有125 Mb的8号染色体!从FASTA文件中提取大量随机子序列可能计算成本相当高昂。
允许容易和快速随机访问的常见计算策略是索引文件。索引文件在生物信息学中是普遍存在的;在未来的章节中,我们将索引基因组,以便它们可以用作比对参考序列,我们将索引包含比对读长的文件,用于快速随机访问。在本节中,我们将介绍一下索引后的FASTA文件是如何允许我们快速轻松地提取子序列的。一般来说,我们经常索引整个基因组,但为了简化本章剩余部分的例子,我们只考虑小鼠基因组的第8号染色体。从本章的GitHub的文件目录中下载 Mus_musculus.GRCm38.75.dna.chromosome.8.fa.gz 文件并用 gunzip 命令解压。

索引压缩的FASTA文件
虽然 samtools faidx 命令确实可以处理压缩的FASTA文件,但是它只适用于BGZF压缩文件(更深入的
话题,我们将在425页“快速访问经过BGZF和Tabix命令索引的制表符分隔的序列文件”进一步讨论)。
我们将使用Samtools对该文件进行索引,它是一种广泛使用的操作SAM和BAM比对格式的工具套件(我们将在第11章介绍更多细节)。你可以通过一个端口或软件包管理器安装 samtools (例如,Mac OS X系统的Homebrew或Ubuntu系统的 apt-get)。 很像Git,Samtools使用子命令来完成的任务。首先,我们需要使用 faidx 子命令索引FASTA文件:
$ samtools faidx Mus_musculus.GRCm38.75.dna.chromosome.8.fa
这个命令将创建一个名为 Mus_musculus.GRCm38.75.dna.chromosome.8.fa.fai 的索引文件。我们在提取子序列时不必担心这个索引文件的细节特征- samtools faidx 命令会注意这些细节的。要访问特定区域的子序列,我们使用 samtools faidx 命令,其中 chromosome: start-end(染色体:起始位置-终止位置)。例如:
$ samtools faidx Mus_musculus.GRCm38.75.dna.chromosome.8.fa 8:123407082-123410744
>8:123407082-123410744
GAGAAAAGCTCCCTTCTTCTCCAGAGTCCCGTCTACCCTGGCTTGGCGAGGGAAAGGAAC
CAGACATATATCAGAGGCAAGTAACCAAGAAGTCTGGAGGTGTTGAGTTTAGGCATGTCT
[...]
一定要注意染色体语法的差异(UCSC的Chr 8与ENSEMBL的8的格式)。如果 samtools faidx 命令没有序列返回,这可能就是原因。
samtools faidx 命令允许一次访问多个区域,因此我们可以这样做:
$ samtools faidx Mus_musculus.GRCm38.75.dna.chromosome.8.fa \
8:123407082-123410744 8:123518835-123536649
>8:123407082-123410744
GAGAAAAGCTCCCTTCTTCTCCAGAGTCCCGTCTACCCTGGCTTGGCGAGGGAAAGGAAC
CAGACATATATCAGAGGCAAGTAACCAAGAAGTCTGGAGGTGTTGAGTTTAGGCATGTCT
[...]
>8:123518835-123536649
TCTCGCGAGGATTTGAGAACCAGCACGGGATCTAGTCGGAGTTGCCAGGAGACCGCGCAG
CCTCCTCTGACCAGCGCCCATCCCGGATTAGTGGAAGTGCTGGACTGCTGGCACCATGGT
[...]
什么让访问索引文件更快?
在第3章(见45页“全能UNIX流程:速度与美貌共存”), 我们讨论了为什么从磁盘读取和写入数据相比存储在内存中的数据非常缓慢。我们可以通过使用指向文件中某些区块位置的索引避免不必要地从磁盘中读取整个文件。在我们使用的FASTA文件的情况下,索引基本上存储量每个序列在文件中的起始位置(以及其他必要的信息)。
当我们查找像8号染色体(123,407,082-123,410,744)这样的区域时,samtools faidx 命令使用索引文件中的信息快速计算那些碱基在文件中的具体位置。然后,使用一个称为文件搜索的操作,程序跳转到文件中的精确位置(称为平移),并开始读取序列。拥有预先计算的文件偏移量和跳转到这些确切位置的能力是快速访问索引文件特定区域的原因。