写在前面:本文为自己在学习过程中看过各类资料后整理做的笔记,纯属方便自己学习以及后期回顾,也希望有幸能帮助到和我一样的小白们,各位大神如果发现有错误即使纠正,我们共同学习!特别感谢所有我阅读过的各类推文的作者,我会附上来源,以示版权及尊重!大家快乐地学习吧!
全基因组关联分析(Genome-Wide Association Study,GWAS)流程

一、准备plink文件
1、准备PED文件
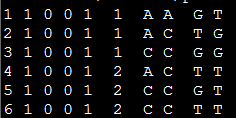
PED文件有六列,六列内容如下:
Family ID
Individual ID
Paternal ID
Maternal ID
Sex (1=male; 2=female; other=unknown)
Phenotype
PED
文件是空格(空格或制表符)分隔的文件。
PED文件长这个样:
2、准备MAP文件
MAP文件有四列,四列内容如下:
chromosome (1-22, X, Y or 0 if unplaced)
rs# or snp identifier
Genetic distance (morgans)
Base-pair position (bp units)
MAP文件长这个样:

3、生成bed、fam、bim、文件
输入命令:
plink --file mydata --out mydata --make-bed
注:plink指的是plink软件,如果软件安装在某个指定的路径的话,前面还要加上路径,比如安装在路径为/your/pathway/的文件夹下,则命令应该为“/your/pathway/plink --file mydata --out mydata --make-bed”
mydata指的是1和2生成的PED和MAP文件名,不需要写.ped和.map后缀
bed:
bim:
fam:
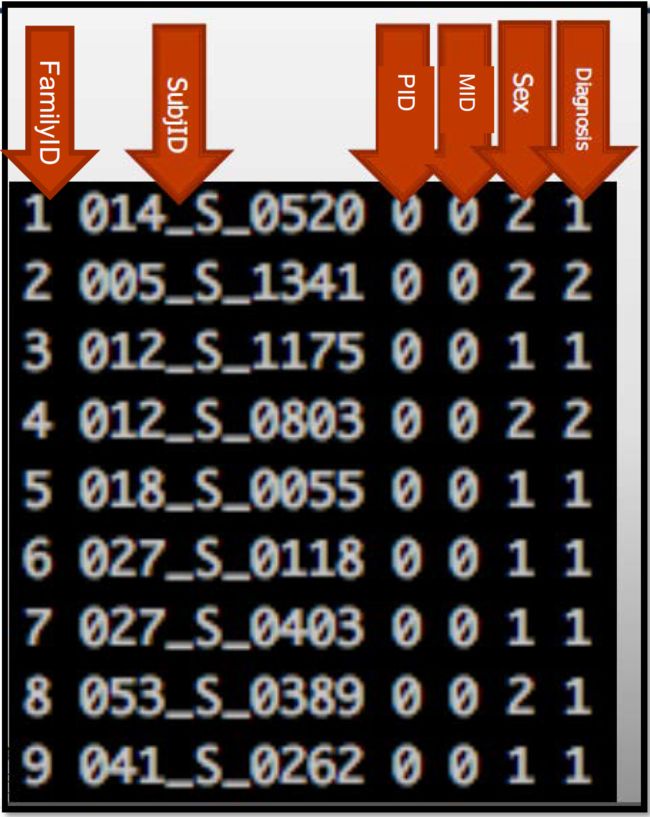
注:有时fam文件最后一列diagnosis会为 -9 或 NA,都表示缺失值
插一步:质控步骤(QC)

可以参照这篇推文,https://www.jianshu.com/p/57c2dbda8a86
填补后质控(Post-imputation quality control)
千人基因组大概有83 million变异位点,经过填补后有许多质量不好的位点,需要过滤掉。
去除MAF = 0的位点
去除MAF<0.01 和 info>0.3的位点。info值用来衡量填充位点的质量,一般较差的位点info <0.15,较好的位点info >0.85。所以过滤阈值一般在0.15-0.85之间。对于同一个位点来说,MAF值越小,info值也越小。可以将MFA值和info值画出柱状图,找到一个比较好的阈值进行过滤。
去除缺失率过多的位点(98%以上)
质控后去除被试命令:
```
plink --bfile test_2kw -remove remove.txt --out 939_test --make-bed --noweb
```
二、准备表型文件(Alternate phenotype files)
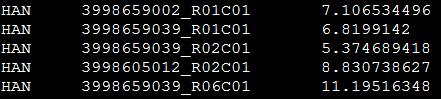
一般表型文件为txt格式,表型文件有三列,分别为:
Family ID
Individual ID
Phenotype
假如有多种表型,第一列和第二列还是Family ID、Individual ID,第三列及以后的每列都是表型,例如以下:
Family ID
Individual ID
Phenotype A
Phenotype B
Phenotype C
……
表型文件长这样:
三、准备协变量文件(Covariate files)
协变量文件同表型文件类似,第一列和第二列是Family ID、Individual ID,第三列及以后的每列都是协变量
Family ID
Individual ID
Covariate A
Covariate B
Covariate C
……
协变量文件长这个样(这里有三个协变量,分别为Sex,Age,temperature):

四、plink进行表型和基因型以及协变量的关联分析
命令如下:
plink --bfile mydata --linear --pheno pheno.txt --mpheno 1 --covar
covar.txt --covar-number 1,2,3 --out mydata –noweb
生成的文件为mydata.assoc.linear
注:“mydata”mydata文件不需要后缀,“--mpheno 1”指的是表型文件的第三列(即第一个表型)
“--covar-number 1,2,3”指的是协变量文件的第三列、第四列、第五列(即第一个、第二个、第三个协变量)
“--linear”指的是用的连续型线性回归,如果表型为二项式(即0、1)类型,则用“--logistic”
五、画曼哈顿图
安装R语言的qqman包,其中的manhattan(),即可画曼哈顿图
https://mp.weixin.qq.com/s/3KN0WHccKv5EbLQUCQVqmw
R包画图
setwd('/Users/mac/Desktop/123') # 设置工作目录
library(qqman) # 载入包
data <- read.table("5filter_result.assoc.linear",header =TRUE) #读取数据
data1 <- data[,c(1,2,3,9)] #按照规则截取列
data2 <- na.omit(data1) # 删除含有NA的整行
par(cex=0.8) #设置点的大小
color_set <- rainbow(9) # 设置颜色集合 建议c("#8DA0CB","#E78AC3","#A6D854","#FFD92F","#E5C494","#66C2A5","#FC8D62")svg(file="manpic.svg", width=12, height=8) # 保存svg格式的图片 设置名字
#manhattan(data2,main="Manhattan Plot",col = color_set) #suggestiveline = FALSE 更加显著
manhattan(data2,main="Manhattan Plot",col = c("#8DA0CB","#E78AC3","#A6D854","#FFD92F","#E5C494","#66C2A5","#FC8D62"),suggestiveline =FALSE,annotatePval =0.01)#suggestiveline = FALSE 更加显著
dev.off() # 保存图片
#par() 显示当前图像参数
str(gwasResults) #zscore beita 值除以standard error 这个值越大 P越小
head(gwasResults) # 看前面几行
tail(data2) #看后面几行
as.data.frame(table(gwasResults$CHR)) # 这个是没根染色体上有多
SNPas.data.frame(table(data2$CHR)) # 这个是没根染色体上有多少SNP
qq(gwasResults$P) # 画qq图
qq(data2$P) # 画qq图
manhattan(gwasResults, annotatePval =0.01) # 这个可以对每根染色体上最高的那个点注释出来
还可以使用haploview画曼哈顿图https://www.jianshu.com/p/609149db6fab
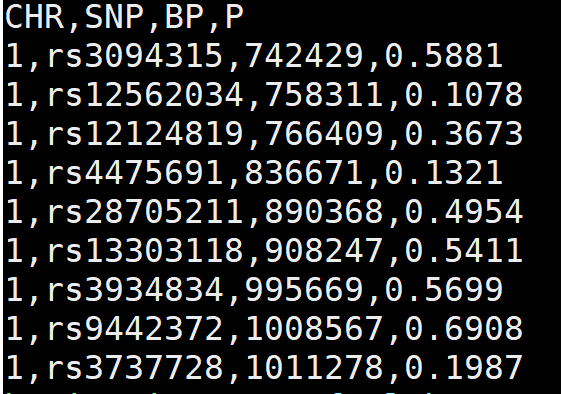
自己入的小坑: assoc.linear文件可以用notepad++打开,但是 .linear不是制表符,需要linux下转换制表符,sed 's/[ ][ ]*/,/g' file.linear > out.csv,sed -i 's/^,//g' out.csv,加了个逗号,最后转成.csv文件,还进行了cut,cut -f 1,2,3,9 file_name > new_file,提取做曼哈顿图时需要的那几列,得到了最终R语言中要做曼哈顿图的文件。我滴个天!哎~
最后需要的文件长这样,好有成就感
其实......不用转格式也可以,后来找到了这个https://www.jianshu.com/p/e914ecb99fcc
直接加载 .linear格式
终于做完了,放一张我自己做的图,啊,虽然还部分有细节问题,但感觉拥有了全世界,啊~


部分还待解决的问题:
1.多个pheno的问题:与单个pheno跑出来的数据大小是一样的,格式也完全一样,p值也只有一列(我以为是多列,每个pheno一个p值),但他们的p值不一样,所以问题来了,计算多表型association时得到的p值是代表什么?多表型一起相关的结果?
2.协变量问题:如果有多个协变量,为什么每个协变量都会跑出来一个p值?我有50万个snp,8个协变量,这样就会出来几百万个p值,都要加载进去做曼哈顿图吗?
3. .assoc.linear文件中最后几行是什么呀,没搞懂

解决问题2:

plink manual中是这样给出的,线性模型的问题,我数学知识有限,讲不太清楚,有大牛看到的话可以同通俗的语言给我留言供大家学习。总之呢,我们关心的是第一个ADD,所以,manual给出了下面的
1.https://www.cnblogs.com/leezx/p/9013615.html
2.http://www.cnblogs.com/chenwenyan/p/6095531.html
3.https://mp.weixin.qq.com/mp/profile_ext?action=home&__biz=MzIwMzQyMTA2NA==&scene=124#wechat_redirect
4.https://mp.weixin.qq.com/s/2Uy9TXbsV267CCnRCa9vQQ
5.GWAS+公共数据库上nature
https://mp.weixin.qq.com/s/TOtYKabcEkkwDVOorB_cYQ
6.找到的视频,不过没有质控
https://www.bilibili.com/video/av35122843/?p=13