文中会对遇到的问题进行解答,图表部分也做了比较详细的说明,对初学者比较友好,当然知道实验部分会让你对数据如何得来的有一个整体的把握,知其然而知其所以然,分析和解释数据起来会更加的得心应手。
本文我首发于微信公众号生信草堂,可在微信公众号生信草堂找到全文(文章标题:16S从实验到数据分析最全流程)和文中示例数据。
导航
本文一共分为三个部分:
不支持页面内跳转
1. 实验部分
2. 建库测序
3. 16S测序数据分析
一. 实验部分:DNA提取与质检
1. DNA提取[1]
- QIAamp fast DNA stool Mini. Ref:51604
- Protocol: Isolation of DNA from stool for Pathogen Detection.
1.1 注意事项:
- ① 如果检测细胞较难溶解(G+菌),可在step3中将水浴温度增加到95℃。
- ② 离心条件:室温,14000rpm。
1.2 准备:
- ① 提前30-40min开启水浴锅。
- ② 在37-70℃中重新溶解Buffer AL和inhibitEX buffer中残留的沉淀。
- ③ 按照说明添加相应体积的酒精于AW1和AW2。
- ④ 加溶液前先混匀。
1.3 实验步骤
实验步骤在原基础上有所修改
- 先将样本在室温下融化10分钟左右。
- 加1.2mL inhibitEX Buffer于2mL离心管中,棉签混匀粪便样本后取180-220mg于2mL的离心管中,vortex直到将样本完全混匀。
-
70℃(可将裂解温度提高到95℃),5min,vortex,15s。 - 离心1min。
- 取上清
550uL到新的1.5mL的EP管中,离心1min。 - 加
30uLproteinase K到新的1.5 mL的EP管中。 - 吸取
400 uL步骤5中的上清液到步骤6中的EP管中。 - 加
400uLbuffer AL, vortex,15s。 - 70℃孵育10min。
- 加
400uL酒精(96-100%),混匀(vortex)。 - 吸取600 uL加入吸附柱,离心1min,弃下管,换新的收集管。
重复步骤11一次。- 加500 uL Buffer AW1, 离心1min,弃下管,换新的收集管。
- 加500 uL Buffer AW2, 离心3min,弃下管,换新的收集管。
- 空管离心3min。
- 将吸附柱转移到新的1.5mL EP管中,加200ul(如果浓度较低可减少体积)Buffer ATE, 室温孵育
5min,离心1min。
2. 质检
仅供参考
DNA浓度:≥10ng/ul[2]
DNA纯度:A260/A280 = 1.8 – 2.0左右
DNA总量:≥300ng[3]
-
DNA完整性:要有明显的基因组主带
 1% 的琼脂糖凝胶,上样量400ng,120v电压跑胶30min
1% 的琼脂糖凝胶,上样量400ng,120v电压跑胶30min这是较好的结果了,有拖带很正常,只要主带明显即可。
3. 总结
- 样本成分复杂,基因组DNA比较容易降解,在保证提取质量的前提下尽量加快前期的样本处理速度。
- 样本反复冻融之后提取的DNA质量较差,保存之前可分装成多管或者一次多提几管备用。
二. 建库测序[4]
- 以下只做一个简单的介绍,仅供参考
流程图

第一轮PCR

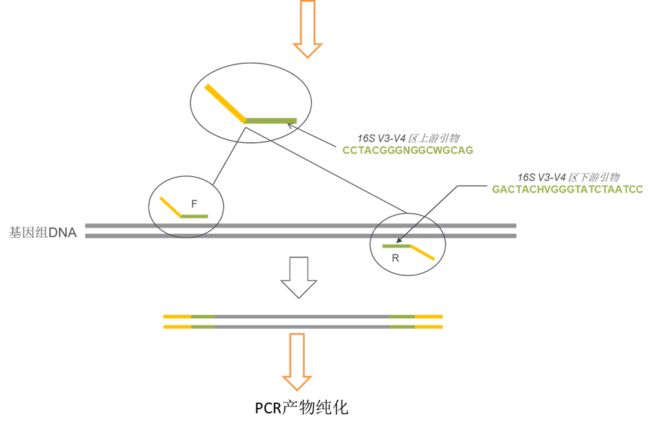
第二轮PCR

RCR产物纯化与文库质检

三. 16S测序数据分析
可在微信公众号:生信草堂,获取示例所用的文件
16S rRNA 基因是编码原核生物核糖体小亚基的基因,长度约1500bp左右,包括9个可变区和10个保守区,保守区序列反映了物种间的亲缘关系,而可变区序列则能反映物种间的差异。经常用于细菌系统发育和分类鉴定,核心是物种分析,包括微生物的种类,不同种类间的相对丰度,不同分组间的物种差异以及系统进化等。一般根据实验目标,设计和样本类型等不同,扩增的区域也会有所不同。
本实例采用的是Illumina
MiSeqV3-V4区的测序数据,长度在460bp左右,为减少运算量等,本例只包含了5个样本,仅供参考。分析流程是在linux上完成,需要有一点linux基础。
16S分析流程图
点击这里查看网页版

1. qiime2安装[5]
1.1 Minicoda软件包管理器安装
# 安装miniconda软件管理器(https://conda.io/miniconda.html):用于安装QIIME2及依赖关系
wget https://repo.continuum.io/miniconda/Miniconda3-latest-MacOSX-x86_64.sh
./Miniconda3-latest-Linux-x86_64.sh
# 如果已经安装过,则升级conda为最新版本
conda update conda
conda install wget
1.2 安装qiime2最新版
wget https://data.qiime2.org/distro/core/qiime2-2018.8-py35-linux-conda.yml
conda env create -n qiime2-2018.8 --file qiime2-2018.8-py35-linux-conda.yml
# OPTIONAL CLEANUP
rm qiime2-2018.8-py35-linux-conda.yml
# 激活工作环境
source activate qiime2-2018.8
# 检查安装是否成功,运行此命令没有报错即安装成功
qiime --help
# 不用时可将其关闭
source deactivate
2. 导入数据分析前的工作(可选)
- 送公司测序返还的数据一般都是拆分过并去除了引物的,可以自己再做一下质检,后续使用dada2分析时也会有碱基质量分布图,所以这步可以不做,自己质检(fastqc)的信息会比较全面。
- 切除引物也只是一个示例,在本例中可以跳过。
2.1 质检
# 公司的给的测序数据一般都是拆分过的,可将单端数据全部合并做质检,也可单独质检
# cat命令合并压缩过的文件会出错,合并之前需要先解压。
gunzip *.gz # 解压
# 第一种方法:合并后质检。
cat *R1* > R1.fastq #合并上游序列,并指定输出文件名为R1.fastq
cat *R2* > R2.fastq #合并下游序列,并指定输出文件名为R2.fastq
# 质检
mkdir qc #创建一个文件夹用于存放质检文件
fastqc -t 2 R1.fastq R2.fastq -o qc # -t --threads,一般有多少个样本用多少线程。-o 指定输出文件存放目录。
# 第二种方法:单独质检后将质检报告合并
mkdir qct #创建一个文件夹用于存放质检文件
fastqc *fastq -t 10 -o qct
pip install multiqc #安装multiqc
multiqc qct/* # 合并报告
- fastqc的结果包括reads各位置的碱基质量值分布、碱基的总体质量值分布、reads各个位置上碱基分布比例、GC含量分布、reads各个位置的N碱基数目、是否含有测序接头序列等。
-
图中横轴是测序序列的碱基,纵轴是质量得分,质量值Q = -10*log10(error P),即20表示1%的错误率,30表示0.1%的错误率。图中每1个箱线图(又称盒须图),都是该位置的所有序列的测序质量的一个统计,分别表示最小值、下四分位数(第25百分位数)、中位数(第50百分位数)、上四分位数(第75百分位数)以及最大值,图中蓝色的细线是各个位置的平均值的连线。
 第一种方法:fasqc碱基质量分布图(R1)
第一种方法:fasqc碱基质量分布图(R1)
 第二种方法:碱基质量分布图
第二种方法:碱基质量分布图
也可在dada2步骤时设置合适的参数去除引物(论坛里建议在使用dada2处理数据之前先去掉引物。参考:https://forum.qiime2.org/t/lost-of-data-with-dada2/1449/5)
2.2 切除引物(本例中的引物已经切除,可跳过)
# 安装与升级cutadapt
pip install --user --upgrade cutadapt
# 将cutadapt添加$PATH环境变量(需管理员权限,没有也没关系,可以跳到下一步)
echo 'PATH="$HOME/.local/bin:$PATH"'>>/etc/profile
# 如无管理员权限,每次使用cutadapt时需指定路径
如:~/.local/bin/cutadapt --help # 如不指定路径则会使用$PATH中的默认版本,老版本不支持多进程
# 创建存放cut的序列的目录
mkdir cut_adapt
# 切除引物序列示例。
~/.local/bin/cutadapt -g forward_primer -e number forward.fastq -o file/R1.fastq -j 0
# cutadapt参数:
-j 0 表示调用所有CPU
-o 指定输出文件目录、文件名。
-g 5’端引物
-a 3’端引物
-e 引物匹配允许错误率,如0.1,0.15等
3. 在qiime2中分析测序数据
3.1 准备工作
# 需要自己写2个文件
# 1.manifest file,将数据放在当前的文件夹下,然后自己写一个绝对路径文件,按以下格式写,#开头的行是注释行会被自动忽略,例如以下命名为为se-33-manifest的文件,也可保存为txt等文件
#absolute filepaths “Fastq manifest” formats *file name: se-33-manifest
sample-id,absolute-filepath,direction
002,/data/shixq/qiime2/002_R1.fastq,forward
002,/data/shixq/qiime2/002_R2.fastq,reverse
017,/data/shixq/qiime2/017_R1.fastq,forward
017,/data/shixq/qiime2/017_R2.fastq,reverse
020,/data/shixq/qiime2/020_R1.fastq,forward
020,/data/shixq/qiime2/020_R2.fastq,reverse
060,/data/shixq/qiime2/060_R1.fastq,forward
060,/data/shixq/qiime2/060_R2.fastq,reverse
091,/data/shixq/qiime2/091_R1.fastq,forward
091,/data/shixq/qiime2/091_R2.fastq,reverse
#另外如果在当前测序数据文件下操作,绝对路径可写为(和上面的写法相等):
sample-id,absolute-filepath,direction
002,$PWD/002_R1.fastq,forward
002,$PWD/002_R2.fastq,reverse
# 2.sample metadata文件,可用EXCEL编辑后保存为制表符分割的txt文件,根据自己的数据做修改。
# 其中q2:types那行可以不写,如果分类是以数字来表示的,如1,2,3,代表不同的分组则需要标注这列对应的q2:types为categorical(分类的),否则会默认为numeric(数字)而报错。
#SampleID subject edu height weight env1 env4
#q2:types categorical categorical categorical categorical categorical
091 subject-1 5 160 50 2 1
020 subject-1 5 160 70 1 3
017 subject-1 2 174 70 1 3
060 subject-2 2 160 70 1 1
002 subject-2 5 174 50 2 1
3.2 激活工作环境
source activate qiime2-2018.8
3.3 导入带质量的双端测序数据
-
质量值体系分为 Phred33 和 Phred 64两种,如下图所示,一般看fastq文件的质量值那行包含!和?(对应ASCII值33和63)等,即为Phred33体系(一般都为Phred33)。
 测序质量值
测序质量值
qiime tools import \
--type 'SampleData[PairedEndSequencesWithQuality]' \
--input-path se-33-manifest \
--output-path paired-end-demux.qza \
--input-format PairedEndFastqManifestPhred33
#可视化文件paired-end-demux.qza
qiime demux summarize \
--i-data paired-end-demux.qza \
--o-visualization paired-end-demux.qzv
-
各样品测序数据柱状分布图,展示不同测序深度下样品数量分布
 pair-end-demux.qzv
pair-end-demux.qzv - 上下游碱基质量分布图

注:
- 生成的.qzv文件可点这里拖拽进网页查看(推荐)
- 或是使用qiime tools view paired-end-demux.qzv 查看
3.4 dada2去燥,合并双端序列
这步也可使用Deblur,这里不做演示,可参考官方文档
# 这步花的时间最长,可使用--p-n-threads 0 参数调用所有CPU,减少运算时间。
注意:需要20bp以上的overlap才能使用dada2拼接,否则会报错。
qiime dada2 denoise-paired \
--i-demultiplexed-seqs paired-end-demux.qza \
--p-trim-left-f 0 \
--p-trim-left-r 0 \
--p-trunc-len-f 270 \
--p-trunc-len-r 250 \
--o-table table.qza \
--o-representative-sequences rep-seqs.qza \
--o-denoising-stats denoising-stats.qza \
--p-n-threads 0 #调用所有CPU
# 可视化denoising stats(qzv文件可在线查看)
qiime metadata tabulate \
--m-input-file denoising-stats.qza \
--o-visualization denoising-stats.qzv
-
展示去除低质量序列、嵌合体、合并等后的序列数
 denoising-stats.qzv
denoising-stats.qzv
3.5 FeatureTable and FeatureData summaries
# Feature表
qiime feature-table summarize \
--i-table table.qza \
--o-visualization table.qzv \
--m-sample-metadata-file sample-metadata.txt
# 代表序列统计
qiime feature-table tabulate-seqs \
--i-data rep-seqs.qza \
--o-visualization rep-seqs.qzv
3.6 建树用于多样性分析
qiime phylogeny align-to-tree-mafft-fasttree \
--i-sequences rep-seqs.qza \
--o-alignment aligned-rep-seqs.qza \
--o-masked-alignment masked-aligned-rep-seqs.qza \
--o-tree unrooted-tree.qza \
--o-rooted-tree rooted-tree.qza
3.7 Alpha多样性分析
# 计算多样性(包括所有常用的Alpha和Beta多样性方法),输入有根树、Feature表、样本重采样深度
# 取样深度看table.qzv文件确定(一般为样本最小的sequence count,或覆盖绝大多数样品的sequence count)
qiime diversity core-metrics-phylogenetic \
--i-phylogeny rooted-tree.qza \
--i-table table.qza \
--p-sampling-depth 55464 \
--m-metadata-file sample-metadata.txt \
--output-dir core-metrics-results
# 输出结果包括多种多样性结果,文件列表和解释如下:
# beta多样性bray_curtis距离矩阵 bray_curtis_distance_matrix.qza
# alpha多样性evenness(均匀度,考虑物种和丰度)指数 evenness_vector.qza
# alpha多样性faith_pd(考虑物种间进化关系)指数 faith_pd_vector.qza
# beta多样性jaccard距离矩阵 jaccard_distance_matrix.qza
# alpha多样性observed_otus(OTU数量)指数 observed_otus_vector.qza
# alpha多样性香农熵(考虑物种和丰度)指数 shannon_vector.qza
# beta多样性unweighted_unifrac距离矩阵,不考虑丰度 unweighted_unifrac_distance_matrix.qza
# beta多样性unweighted_unifrac距离矩阵,考虑丰度 weighted_unifrac_distance_matrix.qza
# 测试分类元数据(sample-metadata)列和alpha多样性数据之间的关联,输入多样性值、sample-medata,输出统计结果
# 统计faith_pd算法Alpha多样性组间差异是否显著
qiime diversity alpha-group-significance \
--i-alpha-diversity core-metrics-results/faith_pd_vector.qza \
--m-metadata-file sample-metadata.txt \
--o-visualization core-metrics-results/faith-pd-group-significance.qzv
# 统计evenness组间差异是否显著
qiime diversity alpha-group-significance \
--i-alpha-diversity core-metrics-results/evenness_vector.qza \
--m-metadata-file sample-metadata.txt \
--o-visualization core-metrics-results/evenness-group-significance.qzv
-
以evenness-group-significance.qzv为例,图中可点Category选择分类方法,查看不同分组下箱线图间的分布与差别。图形下面的表格,详细详述组间比较的显著性和假阳性率统计。
 evenness-group-significance.qzv
evenness-group-significance.qzv
3.8 Beta 多样性分析
# 按subject分组,统计unweighted_unifrace距离的组间是否有显著差异,其他的分组分析类似。
qiime diversity beta-group-significance \
--i-distance-matrix core-metrics-results/unweighted_unifrac_distance_matrix.qza \
--m-metadata-file sample-metadata.txt \
--m-metadata-column subject \
--o-visualization core-metrics-results/unweighted-unifrac-subject-significance.qzv \
--p-pairwise
# 可视化三维展示unweighted-unifrac的主坐标轴分析
qiime emperor plot \
--i-pcoa core-metrics-results/unweighted_unifrac_pcoa_results.qza \
--m-metadata-file sample-metadata.txt \
--p-custom-axes weight \
--o-visualization core-metrics-results/unweighted-unifrac-emperor-weight.qzv
# 可视化三维展示bray-curtis的主坐标轴分析
qiime emperor plot \
--i-pcoa core-metrics-results/unweighted_unifrac_pcoa_results.qza \
--m-metadata-file sample-metadata.txt \
--p-custom-axes height \
--o-visualization core-metrics-results/unweighted-unifrac-emperor-height.qzv
3.9 Alpha rarefaction plotting
# --p-max-depth should be determined by reviewing the “Frequency per sample” information presented in the table.qzv file
that was created above. In general, choosing a value that is somewhere around the median frequency seems to work well.
but you may want to increase that value if the lines in the resulting rarefaction plot don’t appear to be leveling out,
or decrease that value if you seem to be losing many of your samples due to low total frequencies closer to the minimum
sampling depth than the maximum sampling depth.
# --p-max-depth一般取table.qzv文件Frequency per sample的中位数左右
qiime diversity alpha-rarefaction \
--i-table table.qza \
--i-phylogeny rooted-tree.qza \
--p-max-depth 55000 \
--m-metadata-file sample-metadata.txt \
--o-visualization alpha-rarefaction.qzv
- 可视化将有两个图。顶部图是α稀疏图,主要用于确定样品的丰富度是否已被完全观察或测序。如果图中的线在沿x轴的某个采样深度处看起来“平坦化”(即接近零斜率),则表明收集超出该采样深度的其他序列将不可能会有其他的OTU(feature)产生。如果图中的线条没有达到平衡,这可能是因为尚未完全观察到样品的丰富程度(因为收集的序列太少),或者它可能表明在数据中存在大量的测序错误(被误认为是新的多样性)。底部图表示当特征表稀疏到每个采样深度时每个组中保留的样本数。
-
5个样本被分成两组weight,图中显示即两条线,每组的样本数分别为2和3。
 Alpha rarefaction
Alpha rarefaction
3.10 训练分类器
不同实验使用不同的引物有不同的扩增区域,鉴定物种分类的精度就不同,提前的训练可以让分类结果更准确。
提供自己的测序引物序列即可
# 下载数据库文件(greengenes)
wget ftp://greengenes.microbio.me/greengenes_release/gg_13_5/gg_13_8_otus.tar.gz
# 解压
tar -zxvf gg_13_8_otus.tar.gz
# 使用rep_set文件中的99_otus.fasta数据和taxonomy中的99_OTU_taxonomy.txt数据,也可根据需要选择其他相似度。
# 导入参考序列
qiime tools import \
--type 'FeatureData[Sequence]' \
--input-path 99_otus.fasta \
--output-path 99_otus.qza
# 导入物种分类信息
qiime tools import \
--type 'FeatureData[Taxonomy]' \
--input-format HeaderlessTSVTaxonomyFormat \
--input-path 99_otu_taxonomy.txt \
--output-path ref-taxonomy.qza
# Extract reference reads
# 这里不建议指定截取的长度(参考:https://forum.qiime2.org/t/how-can-i-train-classifier-for-paired-end-reads/1512/3)
# Greengenes 13_8 99% OTUs from 341F/805R region of sequences(分类器描述),提供测序的引物序列,截取对应的区域进行比对,达到分类的目的。
qiime feature-classifier extract-reads \
--i-sequences 99_otus.qza \
--p-f-primer CCTACGGGNGGCWGCAG \ #341F引物
--p-r-primer GACTACHVGGGTATCTAATCC \ #805R引物
--o-reads ref-seqs.qza
# Train the classifier(训练分类器)
# 基于筛选的指定区段,生成实验特异的分类器
qiime feature-classifier fit-classifier-naive-bayes \
--i-reference-reads ref-seqs.qza \
--i-reference-taxonomy ref-taxonomy.qza \
--o-classifier Greengenes_13_8_99%_OTUs_341F-805R_classifier.qza
3.11 物种分类
# 物种分类
qiime feature-classifier classify-sklearn \
--i-classifier Greengenes_13_8_99%_OTUs_341F-805R_classifier.qza \
--i-reads rep-seqs.qza \
--o-classification taxonomy.qza
# 结果可视化
qiime metadata tabulate \
--m-input-file taxonomy.qza \
--o-visualization taxonomy.qzv
# 物种分类条形图
qiime taxa barplot \
--i-table table.qza \
--i-taxonomy taxonomy.qza \
--m-metadata-file sample-metadata.txt \
--o-visualization taxa-bar-plots.qzv

-
图中开头字母分别表示:界 (Kingdom)、门(Phylum)、纲 (Class)、目 (Order)、科( Family)、属( Genus)、种 (Species)
 taxa-bar-plots.qzv
taxa-bar-plots.qzv
3.12.1 ANCOM差异度分析
- 差异丰度分析采用ANCOM (analysis of composition of microbiomes),是2015年发布在Microb Ecol Health Dis上的方法,文章称在微生物组方面更专业,但不接受零值(零在二代测序结果表中很常见),用于比较两个或更多群体中微生物组的组成。
- 示例样本较少,没有显著差异,在此不做展示,可参考方法。
# 按subject分组进行差异分析
qiime feature-table filter-samples \
--i-table table.qza \
--m-metadata-file sample-metadata.txt \
--p-where "subject='subject-1'" \
--o-filtered-table subject-1-table.qza
# OTU表添加假count,因为ANCOM不允许有零
qiime composition add-pseudocount \
--i-table subject-1-table.qza \
--o-composition-table comp-subject-1-table.qza
# subject-1 -->weight
qiime composition ancom \
--i-table comp-subject-1-table.qza \
--m-metadata-file sample-metadata.txt \
--m-metadata-column weight \
--o-visualization ancom-subject-1-weight.qzv
3.12.2 按种水平进行差异分析,genus level (i.e. level 6 of the Greengenes taxonomy)
# 按种水平进行合并,统计各种的总reads
qiime taxa collapse \
--i-table subject-1-table.qza \
--i-taxonomy taxonomy.qza \
--p-level 6 \
--o-collapsed-table subject-1-table-l6.qza
# add-pseudocount
qiime composition add-pseudocount \
--i-table subject-1-table-l6.qza \
--o-composition-table comp-subject-1-table-l6.qza
# subject-1 -->weight
qiime composition ancom \
--i-table comp-subject-1-table-l6.qza \
--m-metadata-file sample-metadata.txt \
--m-metadata-column weight \
--o-visualization l6-ancom-subject-1-weight.qzv
refernce:
[1] https://www.qiagen.com/us/shop/sample-technologies/dna/genomic-dna/qiaamp-fast-dna-stool-mini-kit/#resources
[2]、[3] 数值仅供参考,不同的测序公司要求会有不同。
[4]https://support.illumina.com/downloads/16s_metagenomic_sequencing_library_preparation.html
[5] https://docs.qiime2.org/2018.8/install/
[6] https://forum.qiime2.org/t/qiime2-chinese-manual/838
[7] https://forum.qiime2.org/t/lost-of-data-with-dada2/1449/5
[8] https://docs.qiime2.org/2018.8/tutorials/moving-pictures/