看这个之前,可以先看WGCNA的一些理论背景知识
看完整个之后可以去看WGCNA关键模块和hub基因筛选
相似的表达模式可能是
-1 tightly co-regulated
-2 functionally related
-3 members of the same pathway
总体思路流程
-1 准备数据,并进行初步过滤(准备数据,并进行初步过滤)
-2 把表达谱和traits文件载入R并准备成WGCNA需要的格式](#二把表达谱和traits文件载入R并准备成WGCNA需要的格式)
-3 traits热图看有无outlier相当于质量控制(
-4 选择软阈值:为下一步构建网络准备
-5 构建网络(建立模块,并和外部信息构建连接)
方法
两种方法或三种也行:
第一种,一步建网和下面第三种(可以和软件交互,自己设定参数)比较而言,模块自动检测函数内嵌了模块检测的所有步骤,比如自动构建相关性网络,生成cluster树,定义模块为分支,merge相近(相关性大的)模块。输出的是模块颜色,模块特征基因,而这些可以用于后续的分析。同时,user也可以把模块检测的结果可视化智能模块检测的功能有很多参数,并且在作者的例子里,大多数都采取了默认值。我们也试图提供可行的缺省值,但是对于具体的数据分析来说可能不那么合理,因为作者想分析的还不一样。我们鼓励user多读下帮助文件,并改变参数体验一下。
第二种, 作者用的共表达网络用了相对比较少的数据,也就是3600个探针。然而,现在的微阵列测量有的超过50,000个probe表达水平。构建和分析如此大的nodes对一个大型服务器来说也是一种挑战。我们现在演示一种方法,这个也内嵌入了WGCNA包,而这可以让user用这么大的数据进行网络分析。实际上不是使用这么大的数据,软件包会假装电脑硬件不允许使用多于2000个gene进行分析。这个根本的理念是使用two-level聚类。第一,软件使用一个快速的,计算廉价且相对粗糙的聚类方法来预先聚类基因到一个bolck并且数目接近但不会 最大数目2000个gene。然后,对每个block单独执行full网络分析。最后,那些egienggenes高度相关的模块merged到一起。这种方法的优势在于,以比较小的内存就可以运行,而这个问题是在所有标准的桌面电脑中普遍存在的情况,这是计算的加速。这个block或许不是最优的,会导致一些outlying genes被分派到一个不同的module中,而原来在全网络分析可能不在这个模块中。
第三种,逐步进行模块检测可以提供用户和软件相互交互的界面,这非常便利。
作者文件里会展示内嵌到WGCNA里的步步建网的步骤。
1.定义一个权重的邻接矩阵
2.定义这个拓扑重叠矩,基于不相似检测dissTOM
3.构建等级聚类树(average linage)
4.定义树的分支为模块
从芯片原始数据到WGCNA具体步骤
1准备数据,并进行初步过滤
获得CEL数据,并进行初步处理,比如之前的qc,但是这个可以不做,因为后面会有其他甄别方式。在本地磁盘建立文件夹,把所需要的数据命名好全部放进去(命名很重要,后面省却很多麻烦),表达谱文件有人用log2转换的,有人用差异基因,官方文件和专业人士推荐用原始的数据,之前只要进行过滤就可以,把表达差异很小的还有不稳定的数据进行剔除。要知道这个和上面的qc不一样,上面的qc是剔除不合格的芯片,而这一步是剔除芯片中的数据。那么是线剔除芯片还是线处理数据,都可以。先剔除芯片的话会减少点工作量,但对于WGCNA需要的大量样本来说,这点可以忽略。再者专业人士也建议不要剔除芯片除非充足理由要剔除。也就是我们先处理芯片的数据,进行filter.
setwd("D:\\LRWGCNA")
library(affy)
library(limma)
source("http://bioconductor.org/biocLite.R")
source("https://bioconductor.org/biocLite.R")
biocLite("affy")
biocLiate("affyPLM")
biocLite("RcolorBrewer")
biocLite("impute")
biocLite("limma")
biocLite("pheatmap")
biocLite("ggplot2")
Data<-ReadAffy()
sampleNames(Data)
eset.mas5<-mas5(Data)
calls <- mas5calls(Data)
calls<-exprs(calls)
absent <- rowSums(calls == 'A') # how may samples are each gene'absent' in
absent <- which (absent == ncol(calls)) # which genes are 'absent' in all samples
rmaFiltered <- rmaData[-absent,] # filters out the genes 'absent' in all samples
head(rmaFiltered)
write.csv(rmaFiltered,file="wgcna.csv")
准备traits文件,可以提前在excel准备好,然后另存为datTraits.csv
一定注意,第一列sample的名字一定和上面的表达谱文件一致,否则后面读不出来
2 把表达谱和traits文件载入R并准备成WGCNA需要的格式
setwd("D:\\Rworkdirectory\\WGCNA") # Change the text within quotes as necessary. I have downloaded and unzipped the WGCNA folder on my Desktop
# Uploading data into R and formatting it for WGCNA --------------
# This creates an object called "datExpr" that contains the normalized counts file output from DESeq2
datExpr = read.csv("wgcna.csv")
head(datExpr)
# Manipulate file so it matches the format WGCNA needs,处理下符合wgcna的格式
row.names(datExpr) = datExpr$X
datExpr$X = NULL
datExpr = as.data.frame(t(datExpr)) # now samples are rows and genes are columns
dim(datExpr) # 48 samples and 1000 genes (you will have many more genes in reality)
# Run this to check if there are gene outliers
gsg = goodSamplesGenes(datExpr, verbose = 3)
gsg$allOK

这必须里显示是TRUE,否则执行下面代码
#If the last statement returns TRUE, all genes have passed the cuts. If not, we remove the offending genes and samples from the data with the following:
if (!gsg$allOK)
{if (sum(!gsg$goodGenes)>0)
printFlush(paste("Removing genes:", paste(names(datExpr)[!gsg$goodGenes], collapse= ", ")));
if (sum(!gsg$goodSamples)>0)
printFlush(paste("Removing samples:", paste(rownames(datExpr)[!gsg$goodSamples], collapse=", ")))
datExpr= datExpr[gsg$goodSamples, gsg$goodGenes]
}
加载traits文件
#Create an object called "datTraits" that contains your trait data
datTraits = read.csv("LRtraits.csv")
head(datTraits)
#form a data frame analogous to expression data that will hold the clinical traits.
rownames(datTraits) = datTraits$Sample
datTraits$Sample = NULL
table(rownames(datTraits)==rownames(datExpr)) #should return TRUE if datasets align correctly,otherwise your names are out of order

这里必须是TRUE,否则下面无法执行。错误原因应该在命名上。
到现在为止,所有的文件已经上传好了,那就等着进入分析过程。
记得把文件写入本地盘
save(datExpr, datTraits, file="SamplesAndTraits.RData")
#load("SamplesAndTraits.RData")
3 traits热图看有无outlier相当于芯片筛选
# Cluster samples by expression
A = adjacency(t(datExpr),type="signed") # this calculates the whole network connectivity
k = as.numeric(apply(A,2,sum))-1 # standardized connectivity
Z.k = scale(k)
thresholdZ.k = -2.5 # often -2.5
outlierColor = ifelse(Z.k严重离群样本可以剔除,一般建议不要剔除。假如真的想剔除,执行下面代码
# Remove outlying samples
remove.samples = Z.k4 选择软阈值:为下一步构建网络准备
关于软阈值的理论
一般认为,在一个网络中,每个节点的重要性是一样的,而实际上,在很多真实网络中,一个node对应的degree(connectivity,也称为连接度,是与该节点相连的节点数)与这个node出现的概率并不是符合普通的平均分布,而是符合幂分布。这样的网络叫无尺度网络(scale free network)。这种网络有很高异质性,其拓扑结构主要由其中少数具有高连接度的点(hubs,中心点,也叫枢纽)决定。例如对酵母分析蛋白互作网络发现,高degree节点对生存意义更重要。无尺度网络对外部环境有很强的纠错能力。例如一些简单生物即使在苛刻的药物处理和环境干预下仍然能够保持基本的持续生长和繁殖。正是这种特性使得包括代谢网络在内的生物网络对环境有很强的稳定性。但另一方面,这样的特性使得网络本身容易受到攻击,特别是如果网络中的中心点被屏蔽,那么网络可能崩溃,整个网络性质也因此而发生改变。
传统的基因共表达模式的研究主要利用皮尔逊相关系数来衡量基因与基因之间的相关性。近年有研究推荐通过对皮尔逊相关系数设定阈值(硬阈值,与下面的软阈值相对)确定网络的存在。在这种网络中,一个node代表一个基因的表达水平(这点要和cytoscape中的node区分开,cytoscape里的node是基因本身而不是表达水平,里面的连接靠的是已有的基因之间的关系,而不是基因表达水平之间的相关系数),不同情形下,两个节点的相关系数超过预先设定的阈值,说明这两个节点彼此连接。但这样会产生一个问题,如果确定这个阈值。如果人为硬性划一个值,那么可能会丢失一些真实的信息,也就是相关系数0.79和0.81真的差别那么大吗。所以,wgcna对基因间的相关系数进行加权,加权的标准是在每个网络中,基因和基因之间的联系要符合无尺度网络分布。
前面说了,基因共表达网络符合无尺度现象也就是幂律分布。那么wgcna通过对加权系数的选择来获得最符合无尺度网络分布的结果。判断标准符合无尺网络的定义:出现连接度为K的节点的个数的对数log(k)与该节点出现的概率的对数log(p(k))负相关,且相关系数要大于0.8.同时对每一个模块来说,每一个基因的平均连接度应该保证比较高,这样的模块被检测到才有意义。
# Choose a soft threshold power- USE A SUPERCOMPUTER IRL -----------
powers = c(c(1:10), seq(from =10, to=30, by=1)) #choosing a set of soft-thresholding powers
sft = pickSoftThreshold(datExpr, powerVector=powers, verbose =5, networkType="signed") #call network topology analysis function
sizeGrWindow(9,5)
par(mfrow= c(1,2))
cex1=0.9
plot(sft$fitIndices[,1], -sign(sft$fitIndices[,3])*sft$fitIndices[,2], xlab= "Soft Threshold (power)", ylab="Scale Free Topology Model Fit, signed R^2", type= "n", main= paste("Scale independence"))
text(sft$fitIndices[,1], -sign(sft$fitIndices[,3])*sft$fitIndices[,2], labels=powers, cex=cex1, col="red")
#有的论文的是R2大于0.85
abline(h=0.90, col="red")
plot(sft$fitIndices[,1], sft$fitIndices[,5], xlab= "Soft Threshold (power)", ylab="Mean Connectivity", type="n", main = paste("Mean connectivity"))
text(sft$fitIndices[,1], sft$fitIndices[,5], labels=powers, cex=cex1, col="red")
#检验选定的阈值下网络是否逼近scale free
#有错误,无法运行
ADJ1=abs(cor(datExpr,use=”p”))^8
k=as.vector(apply(ADJ1,2,sum,na.rm=T))
sizeGrWindow(10,5)
par(mfrow=c(1,2))
hist(k)
scaleFreePlot(k,main=”Check scale free topology\n)
enableWGCNAThreads()
#用power=1**8****建立邻接矩阵**
softPower = 18
#建立邻接矩阵
adjacency = adjacency(datExpr, power = softPower, type = "signed") #specify network type
#translate the adjacency into topological overlap matrix and calculate the corresponding #dissimilarity:将邻接矩阵转换为拓扑矩阵
TOM = TOMsimilarity(adjacency, TOMType="signed") # specify network type OGM2hours
dissTOM = 1-TOM
# Generate Modules --------------------------------------------------------
# Generate a clustered gene tree以TOM为基础聚类
geneTree = flashClust(as.dist(dissTOM), method="average")
plot(geneTree, xlab="", sub="", main= "Gene Clustering on TOM-based dissimilarity", labels= FALSE, hang=0.04)
#This sets the minimum number of genes to cluster into a module
#动态剪切树法切割模块,选择动态混合切割法
minModuleSize = 30
dynamicMods = cutreeDynamic(dendro= geneTree, distM= dissTOM, deepSplit=2, pamRespectsDendro= FALSE, minClusterSize = minModuleSize)
dynamicColors= labels2colors(dynamicMods)
#合并相似的共表达网络,相当于把上一部筛选得到的modules再进行聚类,使用函数#moduleEigegenes(),确定修剪高度后,使用mergeCloseModules()函数合并modeles
#set a threhold for merging modules. In this example we are not merging so MEDissThres=0.0
MEList= moduleEigengenes(datExpr, colors= dynamicColors,softPower = 18)
MEs= MEList$eigengenes
MEDiss= 1-cor(MEs)
METree= flashClust(as.dist(MEDiss), method= "average")
#plots tree showing how the eigengenes cluster together
plot(METree, main= "Clustering of module eigengenes", xlab= "", sub= "")
#选择相关系数大于0.8的进行合并(王攀用的0.75,下面不是0.2而是0.25)
MEDissThres = 0.2
merge = mergeCloseModules(datExpr, dynamicColors, cutHeight= MEDissThres, verbose =3)
mergedColors = merge$colors
sizeGrWindow(12,9)
mergedMEs = merge$newMEs
save(dynamicMods, MEList, MEs, MEDiss, METree, file= "Network_allSamples_signed_RLDfiltered.RData")
#plot dendrogram with module colors below it
plotDendroAndColors(geneTree, cbind(dynamicColors, mergedColors), c("Dynamic Tree Cut", "Merged dynamic"), dendroLabels= FALSE, hang=0.03, addGuide= TRUE, guideHang=0.05)
moduleColors = mergedColors
colorOrder = c("grey", standardColors(50))
moduleLabels = match(moduleColors, colorOrder)-1
MEs = mergedMEs
save(MEs, moduleLabels, moduleColors, geneTree, file= "Network_allSamples_signed_nomerge_RLDfiltered.RData")
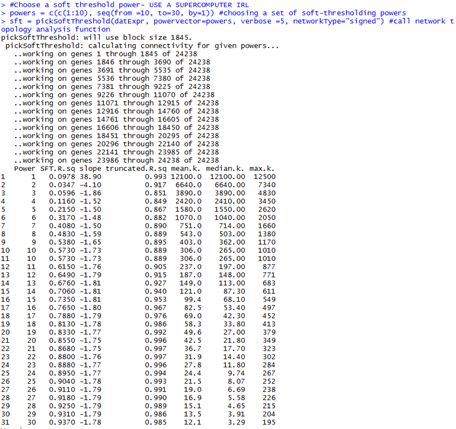
上面这个运行过程有点长大概15mins
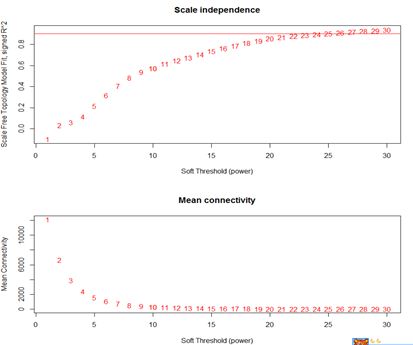
从上面这个图我选择power为24 因为这是阈值到0.90的最小值
5 构建网络(建立模块,并和外部信息构建连接)
具体以下几个步骤
5.1.定义基因共表达的相似矩阵
这个由基因两两之间相关系数的绝对值构成。
2.定义基因网络形成的邻接函数
其实最直接的是对基因间的cor设定硬性阈值,结果要么是有关联要么是无关联。但这会丢失很多信息,前已述及。Wgcna为了克服,提供两种软阈值。两种类似,我们采取其中的幂指数邻接函数方法。就是对每对基因的相关系数进行ß次方幂指数运算,将其加权,其中ß称之为软阈值那么如何选择这个值呢。遵循无尺度网络原则,根据幂律分布,,连接度为K的出现概率p(k)和连接度K的概率cor达到0.85.确定了ß,就是把相关矩阵转变为邻接矩阵。
5.2上述确定了共表达矩阵,那么下一步就确定node间的相异度
基因模块是关联程度高的一组基因组成的。网络中有很多方法描述基因之间的关联程度或共性。Wgcna采取拓扑重叠(topological overlap measure,TOM)计算基因之间的关联程度。这样算的模块有生物意义。把邻接矩阵转换为拓扑矩阵实质是考虑了基因与基因之间通过第三者的间接连接。
5.3.确定基因网络、模块
Wgcna将基因模块定义为高拓扑重叠的一组基因。具体是对基因以TOM为基础的相异度进行层次聚类,建立分层聚类树。一个树叶就是一个基因,不同基因模块便是这棵树的树枝。那如何确定树枝呢,静态剪切树和动态剪切树算法,我们采取动态。
既然共表达的基因很可能受一个转录因子调控,或有同样的功能。那么每个模块和外部信息,比如临床特征等有什么关系吗。我们的例子就是是不是某个模块和某个时间点对应呢?等等。
具体的办法有
-1.计算得到基因模块的特征值,再计算模块的特征向量与关注表型的相关系数
-2.对于分组表型如疾病状态,可以首先定义用t-test计算每个基因在不同组之间的基因差异表达显著性检验p值,并将显著性p值的以10为底的对数值定义为基因显著性(gene significance,GS),再将模块的显著性(module significance,Ms)定义为模块包含的所有基因显著性性的平均值,然后比较MS,一般MS越高,说明这个模块与疾病之间的关联度越高
-3.基因的显著性也可以定义为某个基因表达谱与外部信息的相关性
-4利用基因模块的枢纽基因推测基因模块的成因
# Correlate traits --------------------------------------------------------
#Define number of genes and samples
nGenes = ncol(datExpr)
nSamples = nrow(datExpr)
#Recalculate MEs with color labels
MEs0 = moduleEigengenes(datExpr, moduleColors)$eigengenes
MEs = orderMEs(MEs0)
moduleTraitCor = cor(MEs, datTraits, use= "p")
moduleTraitPvalue = corPvalueStudent(moduleTraitCor, nSamples)
#Print correlation heatmap between modules and traits
textMatrix= paste(signif(moduleTraitCor, 2), "\n(",
signif(moduleTraitPvalue, 1), ")", sep= "")
dim(textMatrix)= dim(moduleTraitCor)
par(mar= c(6, 8.5, 3, 3))
#display the corelation values with a heatmap plot
labeledHeatmap(Matrix= moduleTraitCor,
xLabels= names(datTraits),
yLabels= names(MEs),
ySymbols= names(MEs),
colorLabels= FALSE,
colors= blueWhiteRed(50),
textMatrix= textMatrix,
setStdMargins= FALSE,
cex.text= 0.5,
zlim= c(-1,1),
main= paste("Module-trait relationships"))
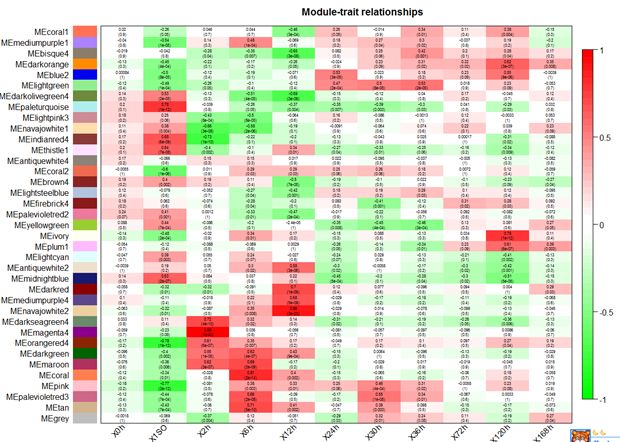
上图可以看出,和SO相关最强的paletruquoise,其次是indianred,thistle,midnightblue,如果想以SO为指标,那可以继续研究这四个模块。同理,2h相关最强的是下方那几个模块,可以深入研究。而2h和6h有重叠。120h有一些正相关和负相关模块,可以深入研究。
由于样本数量有点多,所以会不太清晰。。y轴是模块名,x轴是样本名。。每个模块都跟每个样本有一个空格,空格的上部分是模块与样本的相关系数(代码里面我构建了一个标量矩阵代表样本,然后求了模块和样本的相关系数),空格的下部分是一个p值,我猜是代表相关的显著性吧(不知道理解对不对,但是反正都是挑p值越小的越好)。。由于我不知道这个测试数据的样本信息各个代表咋样的实验,但是如果是有生物学重复实验的话,有人是这样做的:在3个生物学重复中有2个样本在某个模块是显著相关的话,就挑选作为后续研究。
#不同模块基因热图及关键基因的表达
person=cor(datExpr,use = 'p')
corr<-TOM
Colors<-mergedColors
colnames(corr)<-colnames(datExpr)
rownames(corr)<-colnames(datExpr)
names(Colors)<-colnames(datExpr)
colnames(person)<-colnames(datExpr)
rownames(person)<-colnames(datExpr)
umc = unique(mergedColors)
lumc = length(umc)
#change the i to be the number which you want to plot
#改变黄色底的数字可以plot每个模块的图
for (i in c(1:lumc)){
if(umc[i]== "grey"){
next
}
ME=MEs[, paste("ME",umc[3], sep="")]
par(mfrow=c(2,1), mar=c(0.3, 5.5, 3, 2))
plotMat(t(scale(datExpr[,Colors==umc[3]])),nrgcols=30,rlabels=F,rcols=umc[3], main=umc[3], cex.main=3)
par(mar=c(5, 4.2, 0, 0.7))
barplot(ME, col=umc[3], main="", cex.main=2,ylab="eigengene expression",xlab="array sample")
}
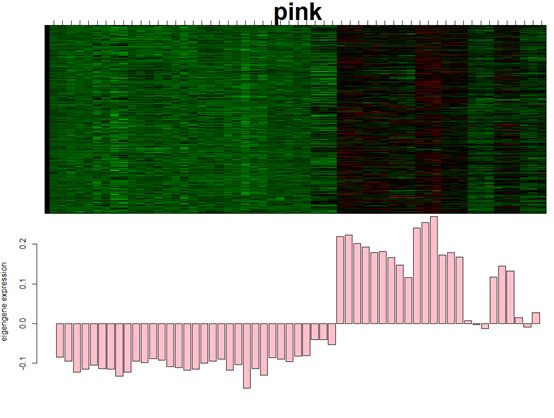
各样本中模块基因与相应ME的表达水平
一个module的基因表达量与该module特征向量图
上图是pink模块所有基因的表达水平,行代表?个模块基因,列代表57个样本。
下面的bar图为对于的样本中pink模块的ME表达水平。可以看出ME的表达水平与整个模块内基因的表达水平高度相关。
简单说也就是pink这个模块在这几个sample中表达最高。
基因共表达网络热图
kME=signedKME(datExpr, mergedMEs, outputColumnName = "kME", corFnc = "cor", corOptions = "use = 'p'")
if (dim(datExpr)[2]>=15000) nSelect=15000 else nSelect=dim(datExpr)[2]
set.seed(1)
select = sample(nGenes, size = nSelect)
selectTOM = dissTOM[select, select]
selectTree = hclust(as.dist(selectTOM), method = "average")
selectColors = moduleColors[select]
plotDiss = selectTOM^7
#the following code will take about mins
#下面这个图会用很长时间,用了3多小时
TOMplot(plotDiss, selectTree, selectColors, main = "Network heatmap plot")
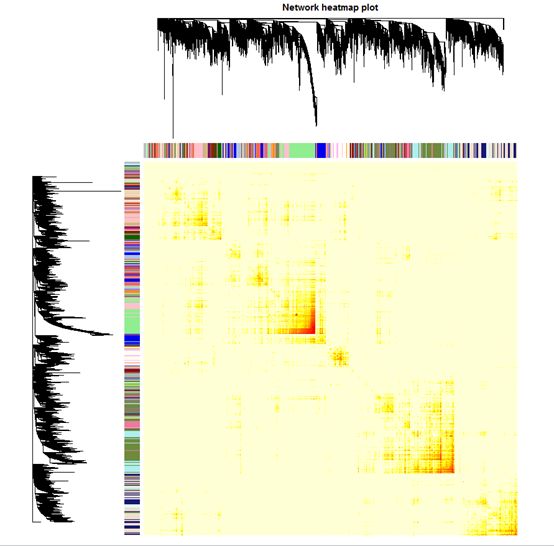
TOM图是指基因和筛选模型的可视化表示。生成TOM图的步骤:1. 生成全基因不相似TOM矩阵,比如1-TOMsimilarityFromExpr(datExpr, power = 6),可以把得到的不相关矩阵加幂,这样绘制的TOM图色彩差异会比较明显。2. 使用TOMplot()绘制TOM图,如上图。图上和图左是全基因系统发育树,不同颜色亮带表示不同的module,每一个亮点表示每个基因与其他基因的相关性强弱(越亮表示相关性越强)。
模块相关性热图
MEs = moduleEigengenes(datExpr, Colors)$eigengenes
MET = orderMEs(MEs)
par(mfrow=c(1,1))
par(mar= c(5, 10.5, 3, 3))
plotEigengeneNetworks(MET, "Eigengene adjacency heatmap", marHeatmap = c(3,4,2,2), plotDendrograms = FALSE, xLabelsAngle = 90)
!](https://upload-images.jianshu.io/upload_images/7976641-aa32e5135f15f7ca.png?imageMogr2/auto-orient/strip%7CimageView2/2/w/1240)
基因显著性和模块身份
研究的一个重要目的是将基因与外部信息的关联程度与基因在相应模块中的重要性联合起来分析,分别被可以由基因显著性和模块身份表示。
模块内部连接度(Intramodular connectivity,IC)用来描述在一个特定的模块中的节点与模块中其他节点的关联程度,可以理解为对模块身份(Module membership,MM)的一种衡量。多数情况下,IC和MM有很高相关性。如下图。但是IC不能衡量某个gene在全局网络中的位置。为了全局网络的层面实现对节点更精确的定位,我们引入模块身份MM的概念。
对某一个基因来说,我们用它在所有样本中的表达值与某个特征向量基因ME表达谱的相关性来衡量这个基因在该模块中的身份。
简单来说就是把每一个关注的基因(比如某模块内的基因),看看它在每个模块的身份值,属于某个模块极值1,否则-1.
与我们关注的某个traits比如2 h相关的枢纽基因
枢纽基因,hub genes,指的是在一个模块中连接度最高的一系列基因。一定程度上他们决定了模块的特征,和全局网络中的枢纽基因相比,模块中的枢纽基因往往更具有生物学意义。在作者的模块truquoise和yellow中,GS和MM具有高度相关性,因此模块中的枢纽基因与临床特征有高相关性。
关于WGCNA hub gene的挑选方法,稍后写出。
最后
ME:module eigengene
IC:intramodular connectivity
GS:gene significance
MM:module membership
WGCNA关键模块和hub基因筛选