之前发现了一个网站分享了很多教程,诸如bash,R,16s扩增子和基因组相关的分析教程,网站叫Happy Belly Bioinformatics。今天来学习分享一下。
1. 主流扩增子分析流程
现如今有很多处理扩增子数据的方法。这些广泛使用的工具或流程主要包括:mothur, usearch, vsearch, Minimum Entropy Decomposition, DADA2, and qiime2 (整合了上述一些工具). 宏观上来看,无论你选择其中的哪个工具或流程处理数据,你将获得非常相似的结果。但是现如今这个领域有一种新趋势,将从传统的OTU方法转向产生ASVs(扩增子序列变异)的单核苷酸解析方法。此外最近 Callahan et al.对这一趋势给出了合理的解释。如果你对这方面还不熟悉,那么你应该知道大多数这方面的专家都会建议你使用一种解决ASVs的方法。可能你想要一个更大的分辨率级别,但最好还是先生成ASVs,然后你总是可以“退出”你的分辨率,在某个阈值上进行聚类,或者通过分类法或其他方法对序列进行分箱。这就是说,聚类OTUs并不是一个一层不变的问题,而是这些OTUs是如何生成的会让你陷入争议之中。请记住,比如usearch/vsearch 这些示例工作流程,这些都不是权威的。
上面一段是翻译的网站上的一段话(翻译依然很烂,请大家去看原文吧),作者很推崇DADA2,认为扩增子分析将由聚OTU转向ASVs。
2.流程中涉及的工具
在此我们将使用 DADA2 作为的数据处理工具. 此外我们还将使用非常方便的 bbtools软件去剔除原始数据中的引物序列。
3. 教程使用的数据
我们将使用已经发表的文章Microbial Communities on Seafloor Basalts at Dorado Outcrop Reflect Level of Alteration and Highlight Global Lithic Clades中的部分数据作为示例。
作者是在2013 MacBook Pro上进行所有分析。我根据自己的条件,数据的下载和预处理在笔记本(i5 2450M,内存8g)Windows 7下的VMware虚拟机中的ubuntu 16系统进行的。DADA2是在Windows 7下的R和Rstudio中进行。
cd ~
curl -L -o dada2_amplicon_ex_workflow.tar.gz https://ndownloader.figshare.com/files/11342996
tar -xvf dada2_amplicon_ex_workflow.tar.gz
rm dada2_amplicon_ex_workflow.tar.gz
cd dada2_amplicon_ex_workflow/
4. 剔除引物序列
数据下载解压后,文件夹中有20个R1,20个R2文件,总共40个fastq文件。先将20个样品名导入一个文件中,如下:
ls *_R1.fq | cut -f1 -d "_" > samples
使用bbtools中BBDukF的剔除引物序列, 需要使用java,同时指定BBDukF的绝对路径调用它。我直接复制粘贴网站上的代码运行,报错了,下面是我修改后科运行的代码。
mh_user@MicrobiomeHelper:~/dada2_amplicon_ex_workflow$ java -ea -Xmx56m -Xms56m -cp /home/mh_user/bbmap/current/ jgi.BBDukF in=B1_sub_R1.fq in2=B1_sub_R2.fq out=B1_sub_R1_trimmed.fq.gz out2=B1_sub_R2_trimmed.fq.gz literal=GTGCCAGCMGCCGCGGTAA,GGACTACHVGGGTWTCTAAT k=10 ordered=t mink=2 ktrim=l rcomp=f minlength=220 maxlength=280 tbo tpe
Executing jgi.BBDukF [in=B1_sub_R1.fq, in2=B1_sub_R2.fq, out=B1_sub_R1_trimmed.fq.gz, out2=B1_sub_R2_trimmed.fq.gz, literal=GTGCCAGCMGCCGCGGTAA,GGACTACHVGGGTWTCTAAT, k=10, ordered=t, mink=2, ktrim=l, rcomp=f, minlength=220, maxlength=280, tbo, tpe]
Version 38.29
Set ORDERED to true
maskMiddle was disabled because useShortKmers=true
3.489 seconds.
Initial:
Memory: max=56m, total=56m, free=37m, used=19m
Added 53 kmers; time: 0.171 seconds.
Memory: max=56m, total=56m, free=36m, used=20m
Input is being processed as paired
Started output streams: 0.671 seconds.
Processing time: 2.183 seconds.
Input: 3342 reads 987870 bases.
KTrimmed: 3323 reads (99.43%) 65086 bases (6.59%)
Trimmed by overlap: 3210 reads (96.05%) 75923 bases (7.69%)
Total Removed: 68 reads (2.03%) 158441 bases (16.04%)
Result: 3274 reads (97.97%) 829429 bases (83.96%)
Time: 3.029 seconds.
Reads Processed: 3342 1.10k reads/sec
Bases Processed: 987k 0.33m bases/sec
运行完产生了两个压缩文件B1_sub_R1_trimmed.fq.gz和B1_sub_R2_trimmed.fq.gz。
上面是对2个文件进行剔除引物序列,现在使用for循环函数对40个进行剔除。
mh_user@MicrobiomeHelper:~/dada2_amplicon_ex_workflow$ for sample in $(cat samples);do echo "On sample: $sample"; java -ea -Xmx56m -Xms56m -cp /home/mh_user/bbmap/current/ jgi.BBDukF in="$sample"_sub_R1.fq in2="$sample"_sub_R2.fq out="$sample"_sub_R1_trimmed.fq.gz out2="$sample"_sub_R2_trimmed.fq.gz literal=GTGCCAGCMGCCGCGGTAA,GGACTACHVGGGTWTCTAAT k=10 ordered=t mink=2 ktrim=l rcomp=f minlength=220 maxlength=280 tbo tpe; done 2> bbduk_primer_trimming_stats.txt
On sample: B1
On sample: B2
On sample: B3
On sample: B4
On sample: BW1
On sample: BW2
On sample: R10
On sample: R11BF
On sample: R11
On sample: R12
On sample: R1A
On sample: R1B
On sample: R2
On sample: R3
On sample: R4
On sample: R5
On sample: R6
On sample: R7
On sample: R8
On sample: R9
#查看一下产出了哪些文件
mh_user@MicrobiomeHelper:~/dada2_amplicon_ex_workflow$ ls
B1_sub_R1.fq R1A_sub_R1.fq
B1_sub_R1_trimmed.fq.gz R1A_sub_R1_trimmed.fq.gz
B1_sub_R2.fq R1A_sub_R2.fq
B1_sub_R2_trimmed.fq.gz R1A_sub_R2_trimmed.fq.gz
B2_sub_R1.fq R1B_sub_R1.fq
B2_sub_R1_trimmed.fq.gz R1B_sub_R1_trimmed.fq.gz
B2_sub_R2.fq R1B_sub_R2.fq
B2_sub_R2_trimmed.fq.gz R1B_sub_R2_trimmed.fq.gz
B3_sub_R1.fq R2_sub_R1.fq
B3_sub_R1_trimmed.fq.gz R2_sub_R1_trimmed.fq.gz
B3_sub_R2.fq R2_sub_R2.fq
B3_sub_R2_trimmed.fq.gz R2_sub_R2_trimmed.fq.gz
B4_sub_R1.fq R3_sub_R1.fq
B4_sub_R1_trimmed.fq.gz R3_sub_R1_trimmed.fq.gz
B4_sub_R2.fq R3_sub_R2.fq
B4_sub_R2_trimmed.fq.gz R3_sub_R2_trimmed.fq.gz
bbduk_primer_trimming_stats.txt R4_sub_R1.fq
BW1_sub_R1.fq R4_sub_R1_trimmed.fq.gz
BW1_sub_R1_trimmed.fq.gz R4_sub_R2.fq
BW1_sub_R2.fq R4_sub_R2_trimmed.fq.gz
BW1_sub_R2_trimmed.fq.gz R5_sub_R1.fq
BW2_sub_R1.fq R5_sub_R1_trimmed.fq.gz
BW2_sub_R1_trimmed.fq.gz R5_sub_R2.fq
BW2_sub_R2.fq R5_sub_R2_trimmed.fq.gz
BW2_sub_R2_trimmed.fq.gz R6_sub_R1.fq
dada2_example.R R6_sub_R1_trimmed.fq.gz
primers.fa R6_sub_R2.fq
R10_sub_R1.fq R6_sub_R2_trimmed.fq.gz
R10_sub_R1_trimmed.fq.gz R7_sub_R1.fq
R10_sub_R2.fq R7_sub_R1_trimmed.fq.gz
R10_sub_R2_trimmed.fq.gz R7_sub_R2.fq
R11BF_sub_R1.fq R7_sub_R2_trimmed.fq.gz
R11BF_sub_R1_trimmed.fq.gz R8_sub_R1.fq
R11BF_sub_R2.fq R8_sub_R1_trimmed.fq.gz
R11BF_sub_R2_trimmed.fq.gz R8_sub_R2.fq
R11_sub_R1.fq R8_sub_R2_trimmed.fq.gz
R11_sub_R1_trimmed.fq.gz R9_sub_R1.fq
R11_sub_R2.fq R9_sub_R1_trimmed.fq.gz
R11_sub_R2_trimmed.fq.gz R9_sub_R2.fq
R12_sub_R1.fq R9_sub_R2_trimmed.fq.gz
R12_sub_R1_trimmed.fq.gz samples
R12_sub_R2.fq silva_nr_v132_train_set.fa.gz
R12_sub_R2_trimmed.fq.gz
查看被剔除掉的序列
mh_user@MicrobiomeHelper:~/dada2_amplicon_ex_workflow$ paste samples <(grep "Total Removed:" bbduk_primer_trimming_stats.txt | cut -f2)
B1 68 reads (2.03%)
B2 46 reads (3.76%)
B3 44 reads (4.17%)
B4 32 reads (3.05%)
BW1 152 reads (3.19%)
BW2 418 reads (3.29%)
R10 772 reads (3.17%)
R11BF 492 reads (2.58%)
R11 710 reads (3.71%)
R12 1138 reads (3.42%)
R1A 1054 reads (4.06%)
R1B 1364 reads (3.98%)
R2 1246 reads (3.41%)
R3 1396 reads (3.74%)
R4 1514 reads (3.81%)
R5 1372 reads (3.53%)
R6 1138 reads (3.61%)
R7 590 reads (3.48%)
R8 960 reads (3.66%)
R9 684 reads (3.67%)
上述结果显示每个样品被剔除序列片段不超过每个样品序列总长的4%。现在我们可以开始进入R使用DADA2了。
5. 使用R中的DADA2处理数据
我是在windows 7系统下安装的R和Rstudio进行的,与原文操作有些不一致,但不影响分析。
5.1 设置工作环境
> library("dada2")
> packageVersion("dada2")
[1] ‘1.9.3’
> setwd("C:\\Users\\Administrator\\Documents\\R_work\\dada2_amplicon_ex_workflow")
> list.files()
[1] "B1_sub_R1.fq" "B1_sub_R1_trimmed.fq.gz"
[3] "B1_sub_R2.fq" "B1_sub_R2_trimmed.fq.gz"
[5] "B2_sub_R1.fq" "B2_sub_R1_trimmed.fq.gz"
[7] "B2_sub_R2.fq" "B2_sub_R2_trimmed.fq.gz"
[9] "B3_sub_R1.fq" "B3_sub_R1_trimmed.fq.gz"
[11] "B3_sub_R2.fq" "B3_sub_R2_trimmed.fq.gz"
[13] "B4_sub_R1.fq" "B4_sub_R1_trimmed.fq.gz"
[15] "B4_sub_R2.fq" "B4_sub_R2_trimmed.fq.gz"
[17] "bbduk_primer_trimming_stats.txt" "BW1_sub_R1.fq"
[19] "BW1_sub_R1_trimmed.fq.gz" "BW1_sub_R2.fq"
[21] "BW1_sub_R2_trimmed.fq.gz" "BW2_sub_R1.fq"
[23] "BW2_sub_R1_trimmed.fq.gz" "BW2_sub_R2.fq"
[25] "BW2_sub_R2_trimmed.fq.gz" "dada2_example.R"
[27] "primers.fa" "R10_sub_R1.fq"
[29] "R10_sub_R1_trimmed.fq.gz" "R10_sub_R2.fq"
[31] "R10_sub_R2_trimmed.fq.gz" "R11_sub_R1.fq"
[33] "R11_sub_R1_trimmed.fq.gz" "R11_sub_R2.fq"
[35] "R11_sub_R2_trimmed.fq.gz" "R11BF_sub_R1.fq"
[37] "R11BF_sub_R1_trimmed.fq.gz" "R11BF_sub_R2.fq"
[39] "R11BF_sub_R2_trimmed.fq.gz" "R12_sub_R1.fq"
[41] "R12_sub_R1_trimmed.fq.gz" "R12_sub_R2.fq"
[43] "R12_sub_R2_trimmed.fq.gz" "R1A_sub_R1.fq"
[45] "R1A_sub_R1_trimmed.fq.gz" "R1A_sub_R2.fq"
[47] "R1A_sub_R2_trimmed.fq.gz" "R1B_sub_R1.fq"
[49] "R1B_sub_R1_trimmed.fq.gz" "R1B_sub_R2.fq"
[51] "R1B_sub_R2_trimmed.fq.gz" "R2_sub_R1.fq"
[53] "R2_sub_R1_trimmed.fq.gz" "R2_sub_R2.fq"
[55] "R2_sub_R2_trimmed.fq.gz" "R3_sub_R1.fq"
[57] "R3_sub_R1_trimmed.fq.gz" "R3_sub_R2.fq"
[59] "R3_sub_R2_trimmed.fq.gz" "R4_sub_R1.fq"
[61] "R4_sub_R1_trimmed.fq.gz" "R4_sub_R2.fq"
[63] "R4_sub_R2_trimmed.fq.gz" "R5_sub_R1.fq"
[65] "R5_sub_R1_trimmed.fq.gz" "R5_sub_R2.fq"
[67] "R5_sub_R2_trimmed.fq.gz" "R6_sub_R1.fq"
[69] "R6_sub_R1_trimmed.fq.gz" "R6_sub_R2.fq"
[71] "R6_sub_R2_trimmed.fq.gz" "R7_sub_R1.fq"
[73] "R7_sub_R1_trimmed.fq.gz" "R7_sub_R2.fq"
[75] "R7_sub_R2_trimmed.fq.gz" "R8_sub_R1.fq"
[77] "R8_sub_R1_trimmed.fq.gz" "R8_sub_R2.fq"
[79] "R8_sub_R2_trimmed.fq.gz" "R9_sub_R1.fq"
[81] "R9_sub_R1_trimmed.fq.gz" "R9_sub_R2.fq"
[83] "R9_sub_R2_trimmed.fq.gz" "samples"
[85] "silva_nr_v132_train_set.fa.gz"
> samples <- scan("samples", what="character")
Read 20 items
> forward_reads <- sort(list.files(pattern="_R1_trimmed.fq.gz"))
> reverse_reads <- sort(list.files(pattern="_R2_trimmed.fq.gz"))
> filtered_forward_reads <- paste0(samples, "_sub_R1_filtered.fq.gz")
> filtered_reverse_reads <- paste0(samples, "_sub_R2_filtered.fq.gz")
5.2 数据过滤
前面使用bbtools剔除引物序列的过程,已经将未完成的或缺失引物的序列剔除,以及那些长度小于220bp或大于280bp的序列剔除。该数据是使用的Illumina MiSeq平台测序产生的 2x300读长的双端序列。上下游引物分别长19和20bp,因此剔除引物后,读长大于280是不正常的。
> plotQualityProfile(forward_reads)
> plotQualityProfile(forward_reads[17:20])
> plotQualityProfile(reverse_reads)
> plotQualityProfile(reverse_reads[17:20])
序列质控图显示20个正向序列质量类似且质量好。

序列质控图显示20个反向序列质量类似但质量明显不如正向序列,这是一个普遍现象,测序仪器先天的缺陷。

Quality Score < 30 的低质量序列部分被切割掉,保证正向序列250bp,反向序列200bp,剩下的高质量的序列,满足后续分析。 Phred讨论了Quality Score 40 和Quality Score 20之间的差异,帮助理解Quality Score。
质控过滤
#在windows下运行的时候提示我没法使用多线程,multithread参数设置为FALSE。
filtered_out <- filterAndTrim(forward_reads, filtered_forward_reads, reverse_reads, filtered_reverse_reads, maxEE=c(2,2), rm.phix=TRUE, multithread=FALSE, minLen=175, truncLen=c(250,200))
> class(filtered_out)
[1] "matrix"
> dim(filtered_out)
[1] 20 2
> filtered_out
reads.in reads.out
B1_sub_R1_trimmed.fq.gz 1637 1533
B2_sub_R1_trimmed.fq.gz 589 541
B3_sub_R1_trimmed.fq.gz 505 472
B4_sub_R1_trimmed.fq.gz 508 485
BW1_sub_R1_trimmed.fq.gz 2304 2153
BW2_sub_R1_trimmed.fq.gz 6151 5744
R10_sub_R1_trimmed.fq.gz 11792 11024
R11_sub_R1_trimmed.fq.gz 9210 8538
R11BF_sub_R1_trimmed.fq.gz 9273 8687
R12_sub_R1_trimmed.fq.gz 16057 14973
R1A_sub_R1_trimmed.fq.gz 12453 11442
R1B_sub_R1_trimmed.fq.gz 16438 15301
R2_sub_R1_trimmed.fq.gz 17670 16351
R3_sub_R1_trimmed.fq.gz 17950 16654
R4_sub_R1_trimmed.fq.gz 19100 17797
R5_sub_R1_trimmed.fq.gz 18745 17522
R6_sub_R1_trimmed.fq.gz 15183 14097
R7_sub_R1_trimmed.fq.gz 8181 7610
R8_sub_R1_trimmed.fq.gz 12622 11781
R9_sub_R1_trimmed.fq.gz 8968 8331
>
质控过滤完后,我们再来看看序列质量。
> plotQualityProfile(filtered_forward_reads)
> plotQualityProfile(filtered_forward_reads[17:20])

> plotQualityProfile(filtered_reverse_reads)
> plotQualityProfile(filtered_reverse_reads[17:20])

6. 生成参数误差模型
> err_forward_reads <- learnErrors(filtered_forward_reads, multithread=TRUE)
47759000 total bases in 191036 reads from 20 samples will be used for learning the error rates.
> err_reverse_reads <- learnErrors(filtered_reverse_reads, multithread=TRUE)
38207200 total bases in 191036 reads from 20 samples will be used for learning the error rates.
序列误差可视化
> plotErrors(err_forward_reads, nominalQ=TRUE)
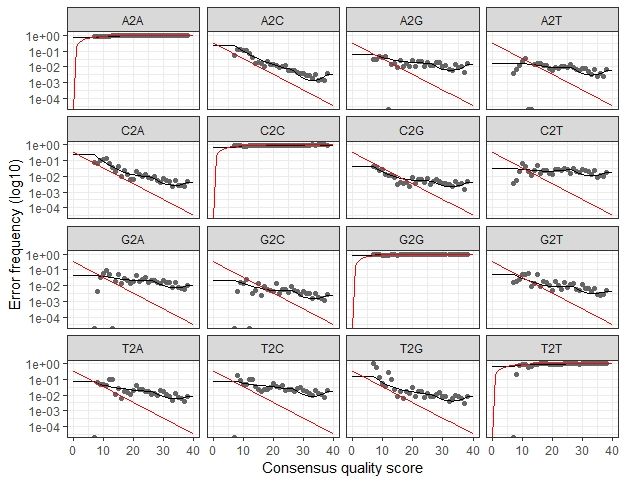
> plotErrors(err_reverse_reads, nominalQ=TRUE)

7. 序列去重复
> derep_forward <- derepFastq(filtered_forward_reads, verbose=TRUE)
Dereplicating sequence entries in Fastq file: B1_sub_R1_filtered.fq.gz
Encountered 566 unique sequences from 1533 total sequences read.
Dereplicating sequence entries in Fastq file: B2_sub_R1_filtered.fq.gz
Encountered 228 unique sequences from 541 total sequences read.
Dereplicating sequence entries in Fastq file: B3_sub_R1_filtered.fq.gz
Encountered 196 unique sequences from 472 total sequences read.
Dereplicating sequence entries in Fastq file: B4_sub_R1_filtered.fq.gz
Encountered 214 unique sequences from 485 total sequences read.
Dereplicating sequence entries in Fastq file: BW1_sub_R1_filtered.fq.gz
Encountered 749 unique sequences from 2153 total sequences read.
Dereplicating sequence entries in Fastq file: BW2_sub_R1_filtered.fq.gz
Encountered 2891 unique sequences from 5744 total sequences read.
Dereplicating sequence entries in Fastq file: R10_sub_R1_filtered.fq.gz
Encountered 5870 unique sequences from 11024 total sequences read.
Dereplicating sequence entries in Fastq file: R11BF_sub_R1_filtered.fq.gz
Encountered 5073 unique sequences from 8538 total sequences read.
Dereplicating sequence entries in Fastq file: R11_sub_R1_filtered.fq.gz
Encountered 3765 unique sequences from 8687 total sequences read.
Dereplicating sequence entries in Fastq file: R12_sub_R1_filtered.fq.gz
Encountered 10140 unique sequences from 14973 total sequences read.
Dereplicating sequence entries in Fastq file: R1A_sub_R1_filtered.fq.gz
Encountered 7236 unique sequences from 11442 total sequences read.
Dereplicating sequence entries in Fastq file: R1B_sub_R1_filtered.fq.gz
Encountered 9819 unique sequences from 15301 total sequences read.
Dereplicating sequence entries in Fastq file: R2_sub_R1_filtered.fq.gz
Encountered 9763 unique sequences from 16351 total sequences read.
Dereplicating sequence entries in Fastq file: R3_sub_R1_filtered.fq.gz
Encountered 10694 unique sequences from 16654 total sequences read.
Dereplicating sequence entries in Fastq file: R4_sub_R1_filtered.fq.gz
Encountered 10683 unique sequences from 17797 total sequences read.
Dereplicating sequence entries in Fastq file: R5_sub_R1_filtered.fq.gz
Encountered 11511 unique sequences from 17522 total sequences read.
Dereplicating sequence entries in Fastq file: R6_sub_R1_filtered.fq.gz
Encountered 9001 unique sequences from 14097 total sequences read.
Dereplicating sequence entries in Fastq file: R7_sub_R1_filtered.fq.gz
Encountered 4598 unique sequences from 7610 total sequences read.
Dereplicating sequence entries in Fastq file: R8_sub_R1_filtered.fq.gz
Encountered 6495 unique sequences from 11781 total sequences read.
Dereplicating sequence entries in Fastq file: R9_sub_R1_filtered.fq.gz
Encountered 4491 unique sequences from 8331 total sequences read.
> names(derep_forward) <- samples
> derep_reverse <- derepFastq(filtered_reverse_reads, verbose=TRUE)
Dereplicating sequence entries in Fastq file: B1_sub_R2_filtered.fq.gz
Encountered 527 unique sequences from 1533 total sequences read.
Dereplicating sequence entries in Fastq file: B2_sub_R2_filtered.fq.gz
Encountered 208 unique sequences from 541 total sequences read.
Dereplicating sequence entries in Fastq file: B3_sub_R2_filtered.fq.gz
Encountered 177 unique sequences from 472 total sequences read.
Dereplicating sequence entries in Fastq file: B4_sub_R2_filtered.fq.gz
Encountered 197 unique sequences from 485 total sequences read.
Dereplicating sequence entries in Fastq file: BW1_sub_R2_filtered.fq.gz
Encountered 679 unique sequences from 2153 total sequences read.
Dereplicating sequence entries in Fastq file: BW2_sub_R2_filtered.fq.gz
Encountered 2593 unique sequences from 5744 total sequences read.
Dereplicating sequence entries in Fastq file: R10_sub_R2_filtered.fq.gz
Encountered 5357 unique sequences from 11024 total sequences read.
Dereplicating sequence entries in Fastq file: R11BF_sub_R2_filtered.fq.gz
Encountered 4764 unique sequences from 8538 total sequences read.
Dereplicating sequence entries in Fastq file: R11_sub_R2_filtered.fq.gz
Encountered 3357 unique sequences from 8687 total sequences read.
Dereplicating sequence entries in Fastq file: R12_sub_R2_filtered.fq.gz
Encountered 9639 unique sequences from 14973 total sequences read.
Dereplicating sequence entries in Fastq file: R1A_sub_R2_filtered.fq.gz
Encountered 6744 unique sequences from 11442 total sequences read.
Dereplicating sequence entries in Fastq file: R1B_sub_R2_filtered.fq.gz
Encountered 9176 unique sequences from 15301 total sequences read.
Dereplicating sequence entries in Fastq file: R2_sub_R2_filtered.fq.gz
Encountered 9138 unique sequences from 16351 total sequences read.
Dereplicating sequence entries in Fastq file: R3_sub_R2_filtered.fq.gz
Encountered 10129 unique sequences from 16654 total sequences read.
Dereplicating sequence entries in Fastq file: R4_sub_R2_filtered.fq.gz
Encountered 9868 unique sequences from 17797 total sequences read.
Dereplicating sequence entries in Fastq file: R5_sub_R2_filtered.fq.gz
Encountered 10884 unique sequences from 17522 total sequences read.
Dereplicating sequence entries in Fastq file: R6_sub_R2_filtered.fq.gz
Encountered 8475 unique sequences from 14097 total sequences read.
Dereplicating sequence entries in Fastq file: R7_sub_R2_filtered.fq.gz
Encountered 4308 unique sequences from 7610 total sequences read.
Dereplicating sequence entries in Fastq file: R8_sub_R2_filtered.fq.gz
Encountered 6000 unique sequences from 11781 total sequences read.
Dereplicating sequence entries in Fastq file: R9_sub_R2_filtered.fq.gz
Encountered 4186 unique sequences from 8331 total sequences read.
> names(derep_reverse) <- samples
8. 推断ASVs
> dada_forward <- dada(derep_forward, err=err_forward_reads, multithread=TRUE, pool="pseudo")
Sample 1 - 1533 reads in 566 unique sequences.
Sample 2 - 541 reads in 228 unique sequences.
Sample 3 - 472 reads in 196 unique sequences.
Sample 4 - 485 reads in 214 unique sequences.
Sample 5 - 2153 reads in 749 unique sequences.
Sample 6 - 5744 reads in 2891 unique sequences.
Sample 7 - 11024 reads in 5870 unique sequences.
Sample 8 - 8538 reads in 5073 unique sequences.
Sample 9 - 8687 reads in 3765 unique sequences.
Sample 10 - 14973 reads in 10140 unique sequences.
Sample 11 - 11442 reads in 7236 unique sequences.
Sample 12 - 15301 reads in 9819 unique sequences.
Sample 13 - 16351 reads in 9763 unique sequences.
Sample 14 - 16654 reads in 10694 unique sequences.
Sample 15 - 17797 reads in 10683 unique sequences.
Sample 16 - 17522 reads in 11511 unique sequences.
Sample 17 - 14097 reads in 9001 unique sequences.
Sample 18 - 7610 reads in 4598 unique sequences.
Sample 19 - 11781 reads in 6495 unique sequences.
Sample 20 - 8331 reads in 4491 unique sequences.
selfConsist step 2
> dada_reverse <- dada(derep_reverse, err=err_reverse_reads, multithread=TRUE, pool="pseudo")
Sample 1 - 1533 reads in 527 unique sequences.
Sample 2 - 541 reads in 208 unique sequences.
Sample 3 - 472 reads in 177 unique sequences.
Sample 4 - 485 reads in 197 unique sequences.
Sample 5 - 2153 reads in 679 unique sequences.
Sample 6 - 5744 reads in 2593 unique sequences.
Sample 7 - 11024 reads in 5357 unique sequences.
Sample 8 - 8538 reads in 4764 unique sequences.
Sample 9 - 8687 reads in 3357 unique sequences.
Sample 10 - 14973 reads in 9639 unique sequences.
Sample 11 - 11442 reads in 6744 unique sequences.
Sample 12 - 15301 reads in 9176 unique sequences.
Sample 13 - 16351 reads in 9138 unique sequences.
Sample 14 - 16654 reads in 10129 unique sequences.
Sample 15 - 17797 reads in 9868 unique sequences.
Sample 16 - 17522 reads in 10884 unique sequences.
Sample 17 - 14097 reads in 8475 unique sequences.
Sample 18 - 7610 reads in 4308 unique sequences.
Sample 19 - 11781 reads in 6000 unique sequences.
Sample 20 - 8331 reads in 4186 unique sequences.
selfConsist step 2
8. 合并双端序列
双端序列合并一般至少需要12bp的重合序列。我们的质控后的正向序列长250bp, 反向序列200。测序V4高变区使用的引物为515f–806r,剔除39bp的引物长度,扩增子片段应该长260bp,因此我们的正反向序列间的重合序列长度可以是190,这一理论值是依据E. coli 16S rRNA gene positions估计的,考虑到实际的生物差异,我们设置重合序列长度为170bp,trimOverhang参数设置为TRUE。
> merged_amplicons <- mergePairs(dada_forward, derep_forward, dada_reverse, derep_reverse, trimOverhang=TRUE, minOverlap=170)
> class(merged_amplicons)
[1] "list"
> length(merged_amplicons)
[1] 20
> names(merged_amplicons)
[1] "B1" "B2" "B3" "B4" "BW1" "BW2" "R10" "R11BF" "R11"
[10] "R12" "R1A" "R1B" "R2" "R3" "R4" "R5" "R6" "R7"
[19] "R8" "R9"
> class(merged_amplicons$B1)
[1] "data.frame"
> names(merged_amplicons$B1)
[1] "sequence" "abundance" "forward" "reverse" "nmatch" "nmismatch"
[7] "nindel" "prefer" "accept"
9.生成序列计数表格
统计双端合并后的数据概况
> seqtab <- makeSequenceTable(merged_amplicons)
> class(seqtab)
[1] "matrix"
> dim(seqtab)
[1] 20 2567
10. Chimera嵌合体的识别和剔除
Chimera(音:奇美拉,魔兽争霸里的双头龙就叫这个名字,游戏没白玩)
> seqtab.nochim <- removeBimeraDenovo(seqtab, multithread=T, verbose=T)
Identified 21 bimeras out of 2567 input sequences.
> sum(seqtab.nochim)/sum(seqtab)
[1] 0.9925714
11. 结果数据的概览
统计每个样品输入序列数,过滤后的序列数,双端合并后的序列数,剔除嵌合体后的序列数等
> getN <- function(x) sum(getUniques(x))
> summary_tab <- data.frame(row.names=samples, dada2_input=filtered_out[,1], filtered=filtered_out[,2], dada_f=sapply(dada_forward, getN), dada_r=sapply(dada_reverse, getN), merged=sapply(merged_amplicons, getN), nonchim=rowSums(seqtab.nochim), total_perc_reads_lost=round(rowSums(seqtab.nochim)/filtered_out[,1]*100, 1))
> summary_tab
dada2_input filtered dada_f dada_r merged nonchim total_perc_reads_lost
B1 1637 1533 1491 1498 1460 1460 89.2
B2 589 541 535 534 533 533 90.5
B3 505 472 465 466 465 465 92.1
B4 508 485 453 457 446 446 87.8
BW1 2304 2153 2109 2122 2076 2076 90.1
BW2 6151 5744 5335 5433 4859 4859 79.0
R10 11792 11024 10266 10465 9574 9403 79.7
R11BF 9210 8538 7648 7879 7120 6981 75.8
R11 9273 8687 8159 8276 7743 7506 80.9
R12 16057 14973 12921 13449 11159 11091 69.1
R1A 12453 11442 10110 10419 9041 9017 72.4
R1B 16438 15301 13513 13964 11699 11653 70.9
R2 17670 16351 14715 15177 13054 12995 73.5
R3 17950 16654 14864 15333 13145 13082 72.9
R4 19100 17797 16703 16940 15278 15212 79.6
R5 18745 17522 15502 16080 13544 13455 71.8
R6 15183 14097 12618 12973 11034 11023 72.6
R7 8181 7610 6782 6982 5931 5919 72.4
R8 12622 11781 10882 11062 10174 10051 79.6
R9 8968 8331 7649 7825 7146 7099 79.2
12. 物种注释
使用silva数据库进行物种注释,也可以选择其他数据库甚至自己构建的个性化数据库。
> taxa <- assignTaxonomy(seqtab.nochim, "silva_nr_v132_train_set.fa.gz", multithread=T, tryRC=T)
13. 从R中导出标准结果文件
asv_seqs <- colnames(seqtab.nochim)
asv_headers <- vector(dim(seqtab.nochim)[2], mode="character")
for (i in 1:dim(seqtab.nochim)[2]) {
asv_headers[i] <- paste(">ASV", i, sep="_")
}
asv_fasta <- c(rbind(asv_headers, asv_seqs))
write(asv_fasta, "ASVs.fa")
asv_tab <- t(seqtab.nochim)
row.names(asv_tab) <- sub(">", "", asv_headers)
write.table(asv_tab, "ASVs_counts.txt", sep="\t", quote=F)
asv_tax <- taxa
row.names(asv_tax) <- sub(">", "", asv_headers)
write.table(asv_tax, "ASVs_taxonomy.txt", sep="\t", quote=F)

至此,我们得到了16s扩增子的ASVs序列,对应的注释信息以及序列数量,下一步需要进行样品的多样性分析,不同样品间物种丰度的差异性分析以及结果的可视化,主要使用R中的 vegan 和 phyloseq包。