1 简介
1.1 scRNA-seq
- 2009年由汤富酬等人首次公开
- 测量每个基因在细胞群中表达水平的分布
- 允许研究在转录组中特定于细胞的变化的非常重要的生物学问题,例如细胞类型识别,细胞反应的异质性,基因表达的随机性,细胞内基因调控网络的推断
- bulk和单细胞RNA-seq之间的主要区别在于,每个测序文库代表单个细胞,而不是细胞群体。因此,必须特别注意比较不同细胞(测序文库)的结果。
- 目前单细胞测序主要由于转录物的起始量较低,在扩增(1 million fold)的时候会产生许多问题。目前,提高转录物捕获效率和降低扩增偏差是研究的活跃领域。可以通过适当的归一化缓解其中的一些问题。
1.2 scRNA-seq的技术
- CEL-seq (Hashimshony et al. 2012)
- CEL-seq2 (Hashimshony et al. 2016)
- Drop-seq (Macosko et al. 2015)
- InDrop-seq (Klein et al. 2015)
- MARS-seq (Jaitin et al. 2014)
- SCRB-seq (Soumillon et al. 2014)
- Seq-well (Gierahn et al. 2017)
- Smart-seq (Picelli et al. 2014)
- Smart-seq2 (Picelli et al. 2014)
- SMARTer
- STRT-seq (Islam et al. 2013)
2 分析流程
2.1 读入数据
本次使用的数据集(Lun et al.2017 )该数据集包含两块416B细胞板(永生的小鼠骨髓祖细胞系),使用Smart-seq2进行处理(Picelli et al,2014)。在制备文库之前,还将spike-in RNA添加到每个细胞的裂解物中。进行高通量测序,并通过计数映射到其外显子区域的读数总数来量化每个基因的表达。类似地,通过计数映射到刺入参考序列的读数的数目来测量每个刺入转录本的量。
首先使用BiocFileCache包下载两个416B细胞板的数据,并将其保存在指定路径中,随后使用read.delim读取并使用SingleCellExperiment转化为单细胞分析中的对象,它可以储存各种信息,包括表达矩阵,基因注释信息,样本分组信息,文库信息和降维信息等绝大部分信息,极大地简化了我们分析时的操作。
library(BiocFileCache)
bfc <- BiocFileCache("raw_data", ask = FALSE)
lun.zip <- bfcrpath(bfc,
file.path("https://www.ebi.ac.uk/arrayexpress/files",
"E-MTAB-5522/E-MTAB-5522.processed.1.zip"))
lun.sdrf <- bfcrpath(bfc,
file.path("https://www.ebi.ac.uk/arrayexpress/files",
"E-MTAB-5522/E-MTAB-5522.sdrf.txt"))
unzip(lun.zip, exdir='E:/simpleSingcell/simpleSingcell')
plate1 <- read.delim(file.path('E:/simpleSingcell/simpleSingcell', "counts_Calero_20160113.tsv"),
header=TRUE, row.names=1, check.names=FALSE)
plate2 <- read.delim(file.path('E:/simpleSingcell/simpleSingcell', "counts_Calero_20160325.tsv"),
header=TRUE, row.names=1, check.names=FALSE)
gene.lengths <- plate1$Length # First column is the gene length.
plate1 <- as.matrix(plate1[,-1]) # Discarding gene length (as it is not a cell).
plate2 <- as.matrix(plate2[,-1])
rbind(Plate1=dim(plate1), Plate2=dim(plate2))
stopifnot(identical(rownames(plate1), rownames(plate2)))
all.counts <- cbind(plate1, plate2)
library(SingleCellExperiment)
sce <- SingleCellExperiment(list(counts=all.counts))
rowData(sce)$GeneLength <- gene.lengths
sce
##查看sce这个SingleCellExperiment,它是一个S4对象,我们可以使用rowData,colData,assay等命令获取对象内的主要信息,seurat中的CreateSeuratObject也是一个S4对象,不过命令稍有不同
#class: SingleCellExperiment
#dim: 46703 192
#metadata(0):
#assays(1): counts
#rownames(46703): ENSMUSG00000102693 ENSMUSG00000064842 ... SIRV7 CBFB-MYH11-mcherry
#rowData names(1): GeneLength
#colnames(192): SLX-9555.N701_S502.C89V9ANXX.s_1.r_1 SLX-9555.N701_S503.C89V9ANXX.s_1.r_1 ...
SLX-11312.N712_S508.H5H5YBBXX.s_8.r_1 SLX-11312.N712_S517.H5H5YBBXX.s_8.r_1
#colData names(0):
#reducedDimNames(0):
#spikeNames(0):
2.2基因注释
由于本例中存在ERCC基因,我们需要将这些基因单独保存在对象中,以便于后续步骤提取;不仅如此,我们还需要获取基因的ensembl id,symbol id,染色体定位等信息,储存在rowData中,将样本的信息保存在colData中。
#首先获取表达矩阵中的ERCC信息
isSpike(sce, "ERCC") <- grepl("^ERCC", rownames(sce))
summary(isSpike(sce, "ERCC"))
#Mode FALSE TRUE
#logical 46611 92
#本例中不仅包含ERCC,作者还添加了SIRV信息,我们将其删掉
is.sirv <- grepl("^SIRV", rownames(sce))
sce <- sce[!is.sirv,]
summary(is.sirv)
#Mode FALSE TRUE
#logical 46696 7
#读取样本信息,并将其添加到colData中,主要包含批次以及实验处理等信息
metadata <- read.delim(lun.sdrf, check.names=FALSE, header=TRUE)
m <- match(colnames(sce), metadata[["Source Name"]]) # Enforcing identical order.
stopifnot(all(!is.na(m))) # Checking that nothing's missing.
metadata <- metadata[m,]
head(colnames(metadata))
#[1] "Source Name" "Comment[ENA_SAMPLE]" "Comment[BioSD_SAMPLE]"
#[4] "Characteristics[organism]" "Characteristics[cell line]" "Characteristics[cell type]"
colData(sce)$Plate <- factor(metadata[["Factor Value[block]"]])
pheno <- metadata[["Factor Value[phenotype]"]]
levels(pheno) <- c("induced", "control")
colData(sce)$Oncogene <- pheno
table(colData(sce)$Oncogene, colData(sce)$Plate)
# 20160113 20160325
# induced 48 48
# control 48 48
#我们可以使用getBMFeatureAnnos使用调取biomaRt包来对基因进行注释
library(scater)
sce <- getBMFeatureAnnos(
sce,
filters = "ensembl_gene_id",
attributes = c(
"ensembl_gene_id",
"external_gene_name",
"chromosome_name",
"start_position",
"end_position"
),
biomart = "ENSEMBL_MART_ENSEMBL",
dataset = "mmusculus_gene_ensembl",
host = "www.ensembl.org"
)
rownames(sce) <- uniquifyFeatureNames(rowData(sce)$ensembl_gene_id, rowData(sce)$external_gene_name)
rowData(sce)$ensembl_gene_id <- rownames(sce)
head(rownames(sce))
#[1] "4933401J01Rik" "Gm26206" "Xkr4" "Gm18956" "Gm37180" "Gm37363"
mito <- which(rowData(sce)$chromosome_name=="MT")
## [1] "Plate" "Oncogene"
## [3] "is_cell_control" "total_features_by_counts"
## [5] "log10_total_features_by_counts" "total_counts"
## [7] "log10_total_counts" "pct_counts_in_top_50_features"
## [9] "pct_counts_in_top_100_features" "pct_counts_in_top_200_features"
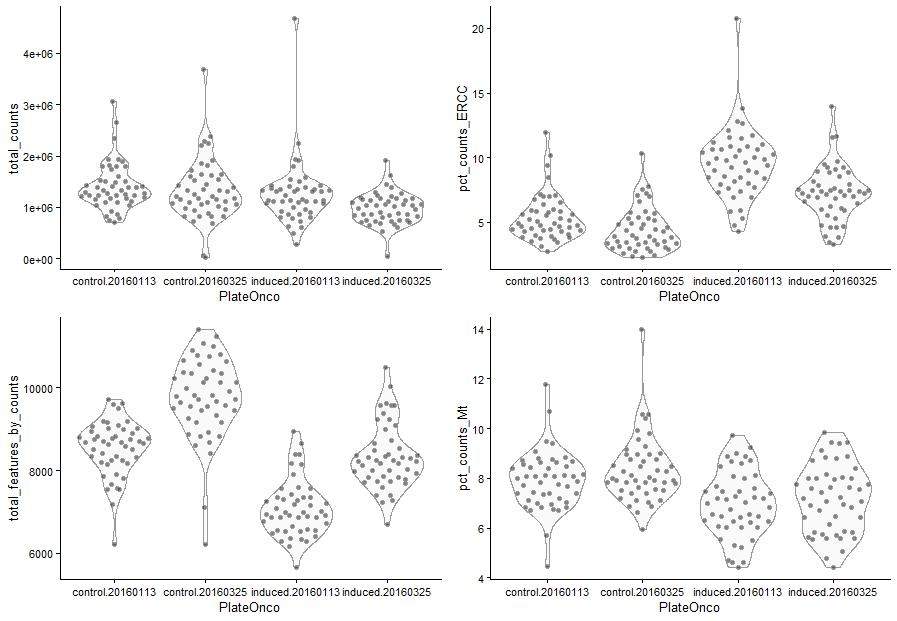
这些指标的分布如图所示,按致癌基因诱导状态和板进行了分层。目的是去除具有低文库大小,少量表达特征和高ERCC(或线粒体)比例的低质量细胞。这样的细胞可以干扰下游分析,例如通过形成使结果解释复杂化的不同簇。
2.3 检测离群值
为这些指标选择一个阈值并不容易,因为它们的值取决于实验方案。不管细胞的质量如何,测序到更大的深度将导致更多的读取和更多表达的特征。同样,在方案中使用更多的刺入RNA将导致更高的ERCC比例。为了获得自适应阈值,我们假设大多数数据集均由高质量单元格组成,并确定对于各种QC指标而言异常的单元格。
离群值是根据与所有单元格中每个指标的中值的中值绝对偏差(MAD)定义的。删除的库大小比中位数库大小大3个MAD的单元格。对数转换可提高小值时的分辨率,尤其是当原始值的MAD等于或大于中值时。还删除了表达基因的对数转化数量低于中位数3 MAD的细胞。
#首先使用calculateQCMetrics函数计算这些质量控制指标,随后做图展示
sce <- calculateQCMetrics(sce, feature_controls=list(Mt=mito))
head(colnames(colData(sce)), 10)
sce$PlateOnco <- paste0(sce$Oncogene, ".", sce$Plate)
multiplot(
plotColData(sce, y="total_counts", x="PlateOnco"),
plotColData(sce, y="total_features_by_counts", x="PlateOnco"),
plotColData(sce, y="pct_counts_ERCC", x="PlateOnco"),
plotColData(sce, y="pct_counts_Mt", x="PlateOnco"),
cols=2)
par(mfrow=c(1,3))
plot(sce$total_features_by_counts, sce$total_counts/1e6, xlab="Number of expressed genes",
ylab="Library size (millions)")
plot(sce$total_features_by_counts, sce$pct_counts_ERCC, xlab="Number of expressed genes",
ylab="ERCC proportion (%)")
plot(sce$total_features_by_counts, sce$pct_counts_Mt, xlab="Number of expressed genes",
ylab="Mitochondrial proportion (%)")

#使用isOutlier计算大于3mad的离群值
libsize.drop <- isOutlier(sce$total_counts, nmads=3, type="lower",
log=TRUE, batch=sce$PlateOnco)
feature.drop <- isOutlier(sce$total_features_by_counts, nmads=3, type="lower",
log=TRUE, batch=sce$PlateOnco)
spike.drop <- isOutlier(sce$pct_counts_ERCC, nmads=3, type="higher",
batch=sce$PlateOnco)
该batch=参数可确保在指定批次的每个水平内识别异常值,防止不感兴趣的因素影响结果。
按上述条件将仅保留高质量单元格。检查每个过滤器除去的细胞数以及保留的细胞总数。删除大量的单元格(> 10%)可能表明数据质量出现整体问题。
keep <- !(libsize.drop | feature.drop | spike.drop)
data.frame(ByLibSize=sum(libsize.drop), ByFeature=sum(feature.drop),
BySpike=sum(spike.drop), Remaining=sum(keep))
## ByLibSize ByFeature BySpike Remaining
## 1 5 4 6 183
然后,将SingleCellExperiment保留高质量细胞,还将原始对象保存到文件中以供以后使用。
sce$PassQC <- keep
saveRDS(sce, file="416B_preQC.rds")
sce <- sce[,keep]
dim(sce)
## [1] 46696 183
还可以使用PCA进行离群值的检测,在运行PCA时,使用detect_outliers函数检测离群值
sce_tmp <- runPCA(sce, use_coldata=TRUE, detect_outliers=TRUE)
table(sce_tmp $outlier)
FALSE TRUE
182 1
还存在一些R包可以供我们质控时使用,cellity使用支持向量机从scRNA-seq中识别低质量细胞。
2.4 细胞周期识别
基于基因表达数据将细胞分类为细胞周期阶段。使用训练数据集,为每对基因计算两个基因之间表达差异的迹象。选择跨细胞周期阶段具有符号变化的对作为标记。然后,可以根据每个标记对的观察符号是否与一个或另一个相一致,将测试数据集中的单元格划分为适当的相,这种方法是在scran包的cyclone函数中实现的。
#使用scran中的cyclone确定细胞周期,目前scran自带人和小鼠的数据可以直接调用
set.seed(100)
library(scran)
mm.pairs <- readRDS(system.file("exdata", "mouse_cycle_markers.rds",
package="scran"))
assignments <- cyclone(sce, mm.pairs, gene.names=rowData(sce)$ensembl_gene_id)
plot(assignments$score$G1, assignments$score$G2M,
xlab="G1 score", ylab="G2/M score", pch=16)
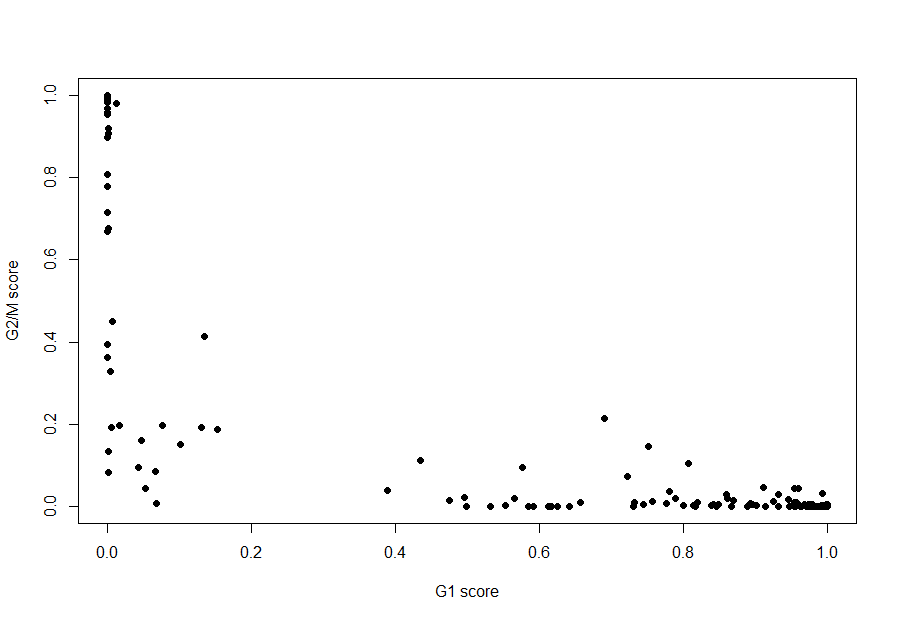
如果G1分数高于0.5且大于G2 / M分数,则将细胞分类为处于G1阶段。如果G2 / M得分高于0.5且高于G1得分,则处于G2 / M阶段;如果两个分数均未超过0.5,则为S阶段。在此,绝大多数细胞被分类为处于G1期。我们将这些分配保存到SingleCellExperiment对象中以供以后使用。
sce$phases <- assignments$phases
table(sce$phases)
##
## G1 G2M S
## 98 62 23
2.5 基因表达质控
检查了表达量最高的基因的身份。通常应以组成型表达的转录本为主,例如核糖体或线粒体蛋白的转录本。如果其他类别的特征与预期的生物学不一致,则可能会引起关注。例如,包含许多ERCC转录本的集合表明在文库制备过程中添加了太多spike-in RNA,而缺少核糖体蛋白和/或它们的假基因则表明存在次佳的比对。
fontsize <- theme(axis.text=element_text(size=12), axis.title=element_text(size=16))
plotHighestExprs(sce, n=50) + fontsize

低丰度基因存在问题,因为零或接近零的计数没有太多信息可用于可靠的统计推断。在涉及假设检验的应用中,这些基因通常不能提供足够的证据来拒绝无效假设,但它们仍会增加多重检验校正的严重性。计数的离散性也可能会干扰统计程序,例如通过破坏连续逼近的准确性。因此,在应用下游方法之前,通常会在许多RNA-seq分析流程中删除低丰度基因。
#首先做一个直方图观察基因表达的分布情况
ave.counts <- calcAverage(sce, use_size_factors=FALSE)
hist(log10(ave.counts), breaks=100, main="", col="grey80",
xlab=expression(Log[10]~"average count"))

可以将最小阈值应用于此值,以滤除表达水平较低的基因。
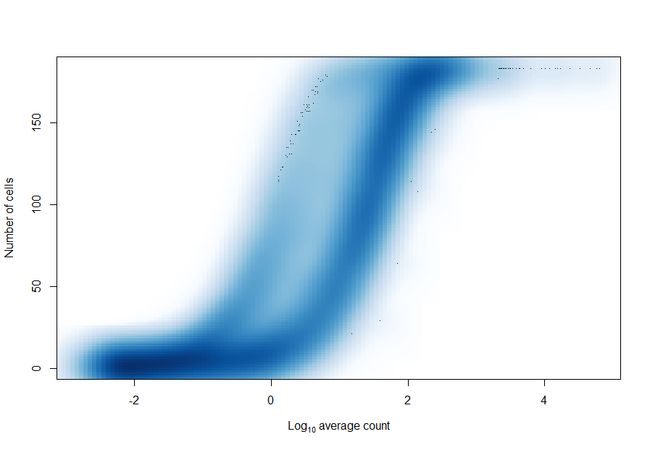
#表达每个基因的细胞数量,这与大多数基因的平均计数密切相关
num.cells <- nexprs(sce, byrow=TRUE)
smoothScatter(log10(ave.counts), num.cells, ylab="Number of cells",
xlab=expression(Log[10]~"average count"))
#在这里我们过滤掉了平均表达量为0的基因,代表在所有细胞中都没有表达的基因。
to.keep <- num.cells > 0
sce <- sce[to.keep,]
summary(to.keep)
## Mode FALSE TRUE
## logical 22833 23863
2.6 标准化表达量
读取count数据受细胞之间捕获效率和测序深度的差异的影响。在进行下游定量分析之前,需要进行标准化以消除这些细胞特异性偏差。这通常是通过假设大多数基因在细胞之间不差异表达(DE)来完成的。假设两个细胞之间跨非DE多数基因的计数大小的任何系统性差异都代表偏差,并通过缩放去除。更具体地说,将计算“size factor”,该大小因子表示每个库中应缩放计数的程度。
#首先快速聚类,如果细胞数量较少可以不使用聚类
set.seed(1000)
clusters <- quickCluster(sce, use.ranks=FALSE, BSPARAM=IrlbaParam())
table(clusters)
#我们使用computeSumFactors计算每个细胞文库大小
sce <- computeSumFactors(sce,min.mean=0.1,clusters=clusters)
summary(sizeFactors(sce))
#以散点图的形式展示size factor与文库之间的相关性
plot(sce$total_counts/1e6, sizeFactors(sce), log="xy",
xlab="Library size (millions)", ylab="Size factor",
col=c("red", "black")[sce$Oncogene], pch=16)
legend("bottomright", col=c("red", "black"), pch=16, cex=1.2,
legend=levels(sce$Oncogene))

由于ERCC基因并不是细胞本身存在的,所以计算size factor时并不适用于ERCC基因,而scater专门为ERCC开发了computeSpikeFactors函数来对ERCC进行标准化
#general.use表示是否应用到全局
sce <- computeSpikeFactors(sce, type="ERCC", general.use=FALSE)
随后使用normalize对表达矩阵进行反卷积标准化,并在同时取对数降维,对数转换是有用的,因为这意味着值的任何差异都直接表示单元格之间表达的对数2倍变化。这通常比覆盖范围的绝对差异更相关,后者需要在总体丰度的背景下进行解释。对数转换还提供了方差稳定化的一些措施,因此具有大方差的高丰度基因不会主导下游分析。数据保存在logcounts中
sce <- normalize(sce)
assayNames(sce)
##[1] "counts" "logcounts"
seurat方法
在seurat中首先使用NormalizeData进行全局缩放归一化方法“ LogNormalize”,该方法将每个单元格的特征表达式测量结果与总表达式进行归一化,再乘以比例因子(默认为10,000),然后对结果进行对数转换。
suerat_object<- NormalizeData(suerat_object, normalization.method = "LogNormalize", scale.factor = 10000)
接下来,我们应用线性变换,这是像PCA这样的降维技术之前的标准预处理步骤。对应ScaleData函数
- 移动每个基因的表达,从而使整个细胞的平均表达为0
- 缩放每个基因的表达,从而使细胞之间的差异为1
- 此步骤在下游分析中具有相同的权重,因此高表达的基因不会占主导地位
- 结果存储在 pbmc[["RNA"]]@scale.data
all.genes <- rownames(suerat_object)
suerat_object<- ScaleData(suerat_object, features = all.genes)
2.7 方差建模筛选HVG
真正的生物异质性或无趣的技术噪音可能会驱动跨基因观察到的表达值的差异。为了区分这两种可能性,我们需要对每个基因的表达值差异的技术成分进行建模。我们使用一组ERCC转录本来完成,这些转录本以相同的数量添加到每个单元格中。因此,ERCC转录本不应表现出生物学变异性,即其表达的任何变异都应是技术来源。
我们使用该trendVar函数将均值相关趋势拟合为ERCC转录本的对数表达值的方差。block参数将对设定批次,以确保各板之间的技术差异不会扩大差异。给定一个基因的平均丰度,然后将趋势的拟合值用作该基因的技术成分的估计值。最后,通过decomposeVar函数从每个基因的总方差中减去技术成分,来计算方差的生物学成分。
var.fit <- trendVar(sce, parametric=TRUE, block=sce$Plate,
loess.args=list(span=0.3))
var.out <- decomposeVar(sce, var.fit)
head(var.out)
## DataFrame with 6 rows and 6 columns
## mean total
##
## ENSMUSG00000103377 0.00807160215928894 0.011921865486065
## ENSMUSG00000103147 0.0346526072192529 0.0722196162535234
## ENSMUSG00000103161 0.00519472222570747 0.00493857699521053
## ENSMUSG00000102331 0.018666093059853 0.032923591860573
## ENSMUSG00000102948 0.059057000132083 0.0881371257735823
## Rp1 0.0970243712569606 0.45233813529556
## bio tech p.value
##
## ENSMUSG00000103377 -0.0238255786088717 0.0357474440949367 1
## ENSMUSG00000103147 -0.0812680860584481 0.153487702311972 0.999999999992144
## ENSMUSG00000103161 -0.0180705438722202 0.0230091208674307 1
## ENSMUSG00000102331 -0.0497487337065681 0.082672325567141 0.999999999998056
## ENSMUSG00000102948 -0.173441452696662 0.261578578470245 1
## Rp1 0.0226096722909625 0.429728463004597 0.0354980966384924
## FDR
##
## ENSMUSG00000103377 1
## ENSMUSG00000103147 1
## ENSMUSG00000103161 1
## ENSMUSG00000102331 1
## ENSMUSG00000102948 1
## Rp1 0.153727758280855
随后将拟合出来的结果进行展示
plot(var.out$mean, var.out$total, pch=16, cex=0.6, xlab="Mean log-expression",
ylab="Variance of log-expression")
curve(var.fit$trend(x), col="dodgerblue", lwd=2, add=TRUE)
cur.spike <- isSpike(sce)
points(var.out$mean[cur.spike], var.out$total[cur.spike], col="red", pch=16)

随后检查具有最大生物学成分的基因的表达值分布。这样可确保方差估计不受一个或两个异常值的影响。
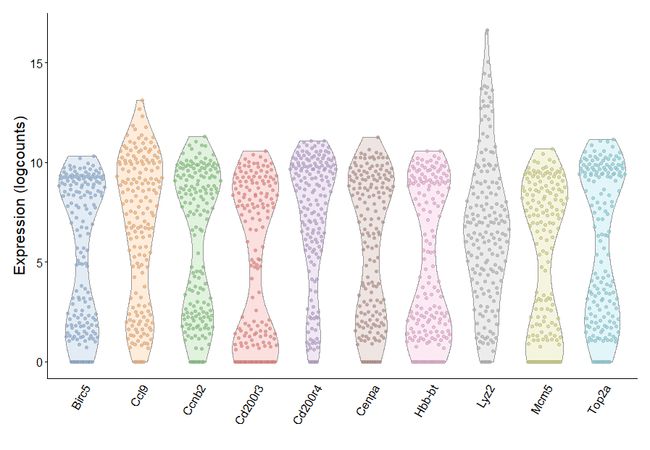
chosen.genes <- order(var.out$bio, decreasing=TRUE)[1:10]
plotExpression(sce, features=rownames(var.out)[chosen.genes]) + fontsize
- 当我们的测序样本中没有添加spike-in基因的时候,我们就需要使用全部基因来建立拟合趋势。
new.trend <- makeTechTrend(x=sce)
fit <- trendVar(sce, use.spikes=FALSE, loess.args=list(span=0.05))
plot(fit$mean, fit$var, pch=16)
curve(fit$trend(x), col="dodgerblue", add=TRUE)
curve(new.trend(x), col="red", add=TRUE)
#使用全部基因拟合出来的趋势寻找HVG
fit$trend <- new.trend # overwrite trend.
dec <- decomposeVar(fit=fit) # use per-gene variance estimates in 'fit'.
top.dec <- dec[order(dec$bio, decreasing=TRUE),]
head(top.dec)
2.8 去除批次效应
limma
如前所述,数据是在两个板上收集的。板之间的加工中不可控的微小差异会导致批次效应,即不同板上细胞之间表达的系统差异。这种差异我们并不关心,可以通过removeBatchEffect实现。这消除了批次的作用,同时考虑了致癌基因诱导的作用。
library(limma)
assay(sce, "corrected") <- removeBatchEffect(logcounts(sce),
design=model.matrix(~sce$Oncogene), batch=sce$Plate)
assayNames(sce)
## [1] "counts" "logcounts" "corrected"
当我们的数据类型相对简单的时候可以使用removeBatchEffect,它是假定细胞群的组成在各批次之间是已知的或相同的。如果我们想要合并更加复杂的数据集的时候,由于scRNA-seq的特性,有一些方法专门用于处理单细胞测序的批次效应,例如MNN方法,seurat的CCA方法等。
MNN去除批次效应
使用fastMNN函数应用于消除批次效应。为了减少计算工作量和技术噪声,将所有数据投影到低维空间中。然后在该低维空间中执行MNN的识别和校正向量的计算。该函数返回一个SingleCellExperiment包含低维校正值的对象,用于下游分析(如聚类或可视化)。
#首先对两个样本的基因名取交集
universe <- intersect(rownames(sce1), rownames(sce2))
#计算两个样本平均生物学差异
mean.bio <- (sce1[universe,"bio"] + sce2[universe,"bio"])/2
chosen <- universe[mean.bio > 0]
length(chosen)
重新缩放每个批次,以调整批次之间测序深度的差异。multiBatchNorm在调整大小因数以实现SingleCellExperiment对象之间覆盖率的系统差异之后,该函数将重新计算对数归一化的表达式值。(请注意,先前计算的大小的因素仅除去细胞之间的偏差内的单个批次)。这通过去除的批次之间的技术差异一方面提高了校正的质量。
rescaled <- batchelor::multiBatchNorm(
sce.gse85241[universe,],
sce.gse81076[universe,]
)
rescaled.gse85241 <- rescaled[[1]]
rescaled.gse81076 <- rescaled[[2]]
set.seed(100)
unc.gse81076 <- logcounts(rescaled.gse81076)[chosen,]
unc.gse85241 <- logcounts(rescaled.gse85241)[chosen,]
mnn.out <- batchelor::fastMNN(
GSE81076=unc.gse81076, GSE85241=unc.gse85241,
k=20, d=50, BSPARAM=IrlbaParam(deferred=TRUE)
)
mnn.out
2.8 降维
我们使用denoisePCA进行PCA线性降维,使用technical参数设定技术噪音,denoisePCA可以同时进行PC的筛选,删除不重要的PC以便于后续的分析。
sce <- denoisePCA(sce, technical=var.out, assay.type="corrected")
dim(reducedDim(sce, "PCA"))
## [1] 183 24
在降维之后,我们可以在低维空间对结果进行可视化。
#查看前三个PC的情况
plotReducedDim(sce, use_dimred="PCA", ncomponents=3,
colour_by="Oncogene") + fontsize
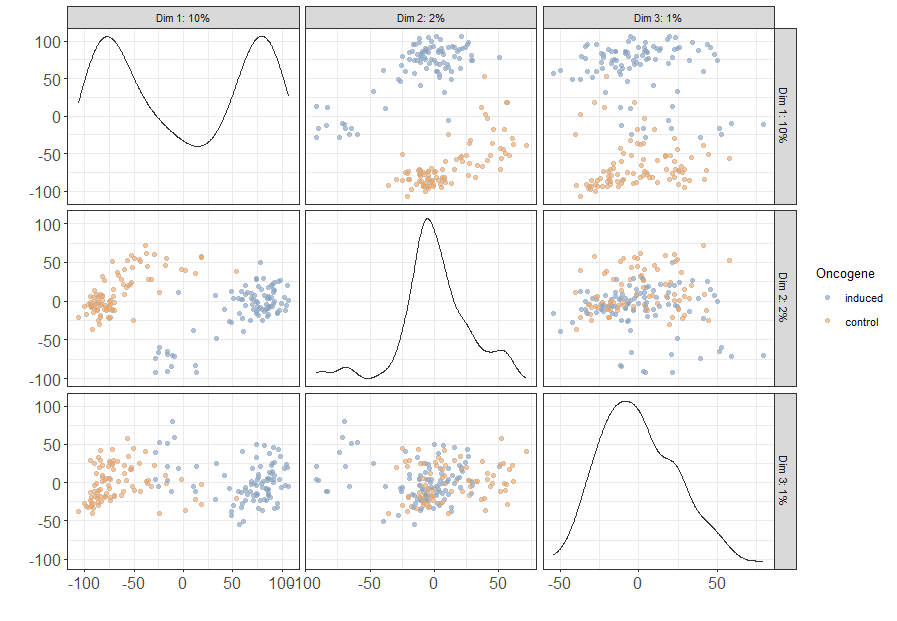
相比之下,我们使用批次信息对细胞进行分组,可以发现并没有明显的细胞分离,这证明我们的批次更正步骤已成功。
plotReducedDim(sce, use_dimred="PCA", ncomponents=3,
colour_by="Plate") + fontsize

请注意,plotReducedDim将使用已经存储在主成分分析sce的denoisePCA。这使我们能够快速生成具有不同美感的新图,而无需重复整个PCA计算。同样,plotPCA将使用现有结果(如果可用),否则将重新计算。用户应设置rerun=TRUE为在存在现有结果的情况下强制重新计算PC。
- 在PCA的基础上,我们继续使用t-SNE进行非线性降维, t- SNE可以比PCA更好地分离更多种群体中的细胞。这是因为前者可以直接捕获高维空间中的非线性关系,而后者必须在线性轴上表示它们。但是,这种改进是以更多的计算工作为代价的。
set.seed(100)
out5 <- plotTSNE(sce, run_args=list(use_dimred="PCA", perplexity=5),
colour_by="Oncogene") + fontsize + ggtitle("Perplexity = 5")
set.seed(100)
out10 <- plotTSNE(sce, run_args=list(use_dimred="PCA", perplexity=10),
colour_by="Oncogene") + fontsize + ggtitle("Perplexity = 10")
set.seed(100)
out20 <- plotTSNE(sce, run_args=list(use_dimred="PCA", perplexity=20),
colour_by="Oncogene") + fontsize + ggtitle("Perplexity = 20")
multiplot(out5, out10, out20, cols=3)
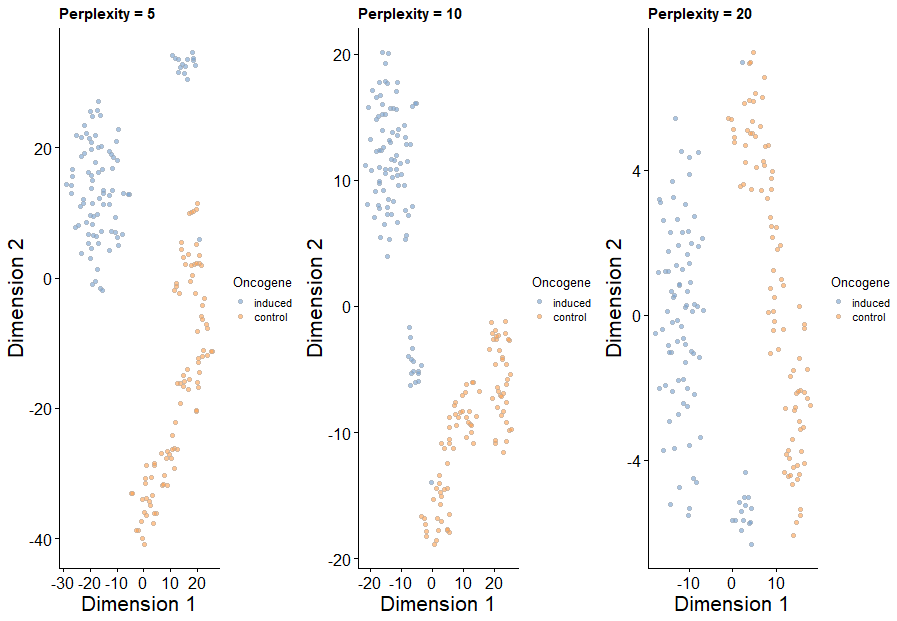
t-SNE是一种随机方法,因此用户应多次运行该算法以确保结果具有代表性。脚本应通过set.seed设置一个种子,以确保所选结果可再现。还建议测试perplexity参数的不同设置,因为这会影响低维空间中点的分布。
在这里,我们runTSNE以20的perplexity进行调用,以将t-SNE结果存储在SingleCellExperiment对象中。这避免了每当我们想使用创建新图时都重复计算plotTSNE,因为将使用存储的结果。同样,用户可以设置rerun=TRUE为在存在存储结果的情况下强制重新计算。
set.seed(100)
sce <- runTSNE(sce, use_dimred="PCA", perplexity=20)
reducedDimNames(sce)
## [1] "PCA" "TSNE"
2.9 细胞聚类
使用层次聚类划分亚群
去噪的对数表达值用于将细胞聚类为推定的亚群。具体来说,我们使用Ward.D2对单元之间的欧式距离进行层次聚类,以使每个聚类中的总方差最小。这将产生一个树状图,该树状图将跨所选基因具有相似表达模式的细胞分组在一起。
pcs <- reducedDim(sce, "PCA")
my.dist <- dist(pcs)
my.tree <- hclust(my.dist, method="ward.D2")
通过对树状图应用动态树切割来明确定义聚类。这利用了树状图中的分支形状来完善聚类定义,并且比cutree复杂树状图更合适。通过在中手动指定cutHeight,cutreeDynamic函数可以更好地控制亚群的划分。我们还设置minClusterSize了一个低于默认值20的值,以避免划分虚假的小亚群。
library(dynamicTreeCut)
my.clusters <- unname(cutreeDynamic(my.tree, distM=as.matrix(my.dist),
minClusterSize=10, verbose=0))
随后使用不同条件对聚类结果进行检查
#使用批次信息
table(my.clusters, sce$Plate)
#使用实验处理信息
table(my.clusters, sce$Oncogene)
在t-SNE图中展示亚群的划分,通常比较接近的细胞会被划分在同一亚群中。
sce$cluster <- factor(my.clusters)
plotTSNE(sce, colour_by="cluster") + fontsize

在聚类之后,我们使用轮廓宽度检查聚类的分离性。理想情况下,每个亚群将包含许多具有较大正宽度的单元,这表明该群集与其他群集完全分开。
library(cluster)
clust.col <- scater:::.get_palette("tableau10medium") # hidden scater colours
sil <- silhouette(my.clusters, dist = my.dist)
sil.cols <- clust.col[ifelse(sil[,3] > 0, sil[,1], sil[,2])]
sil.cols <- sil.cols[order(-sil[,1], sil[,3])]
plot(sil, main = paste(length(unique(my.clusters)), "clusters"),
border=sil.cols, col=sil.cols, do.col.sort=FALSE)

使用图聚类划分亚群
我们建立一个共享的最近邻图,并使用Walktrap算法来识别聚类。
snn.gr <- buildSNNGraph(sce, use.dimred="PCA")
clusters <- igraph::cluster_walktrap(snn.gr)
sce$Cluster <- factor(clusters$membership)
table(sce$Cluster)
## 1 2 3 4

##53 13 37 80
我们查看观察到的边缘权重与预期边缘权重之比,以确认群集是模块化的。(我们没有看模块性评分本身,因为模块性评分在群集之间的变化幅度不同,并且难以解释。)下图指出,大多数群集都很好地分离开了,几乎没有强烈的相关亚群。
cluster.mod <- clusterModularity(snn.gr, sce$Cluster, get.values=TRUE)
log.ratio <- log2(cluster.mod$observed/cluster.mod$expected + 1)
library(pheatmap)
pheatmap(log.ratio, cluster_rows=FALSE, cluster_cols=FALSE,
color=colorRampPalette(c("white", "blue"))(100))

随后在t-SNE中检查聚类情况
plotTSNE(sce, colour_by="Cluster")

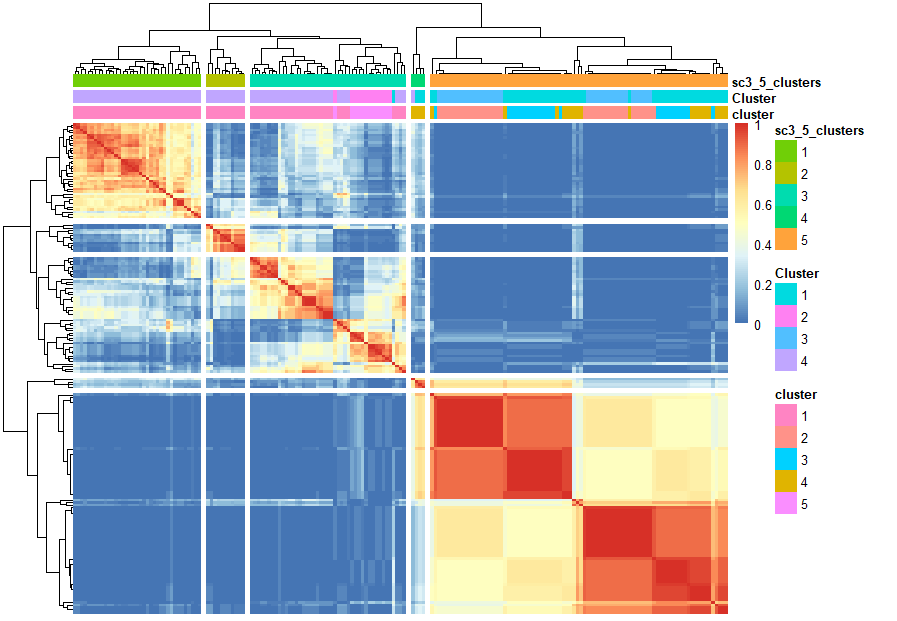
使用一致性聚类对亚群进行划分
让我们SC3对数据进行聚类。SC3的优点是它可以直接摄取SingleCellExperiment对象,当我们不清楚聚类条目K时,SC3可以估计多个群集
library(SC3) # BiocManager::install('SC3')
sce <- sc3_estimate_k(sce) # 先预估一下聚类亚群
sce@metadata$sc3$k_estimation # 预估13个亚群
rowData(sce)$feature_symbol <- rownames(rowData(sce))
# 接下来正式运行,kn参数表示的预估聚类数
# 我们这里自定义为5组
kn <- 5
start=Sys.time()
sce <- sc3(sce, ks = kn, biology = TRUE)
end=Sys.time()
(dur=end-start)
# 会将聚类结果放入表型信息(sce@colData)中去,默认叫sc3_cluster,这里人为改个名称
sc3_cluster="sc3_5_clusters"
# 最后进行可视化比较之前tSNE的Kmeans聚类和SC3的聚类的一致性
sc3_plot_consensus(sce, k = kn, show_pdata = c("cluster",'sc3_5_clusters'))
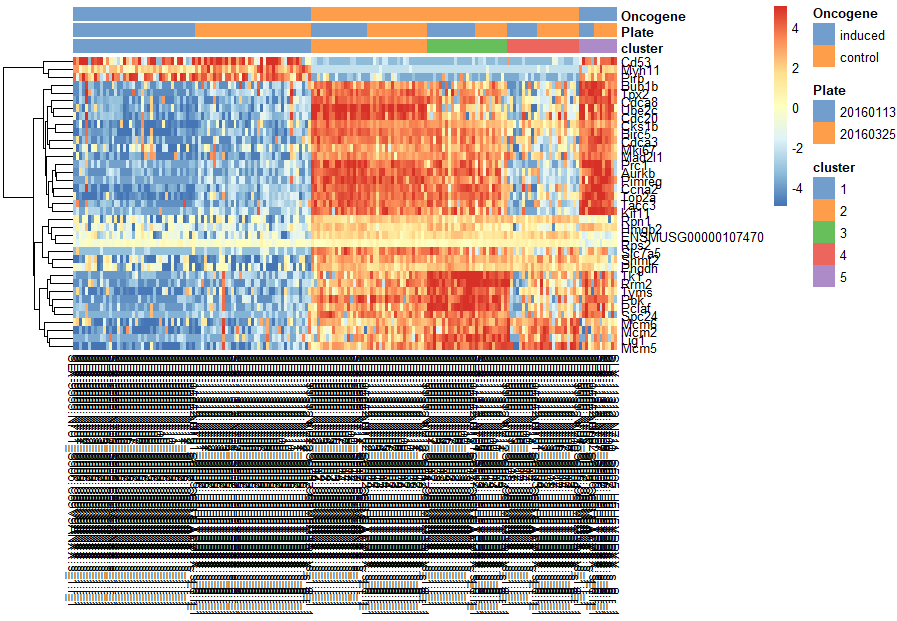
2.10 Marker基因的筛选
我们使用findMarkers来寻找每个亚群中的Marker基因
top.markers <- rownames(marker.set)[marker.set$Top <= 10]
plotHeatmap(sce, features=top.markers, columns=order(sce$cluster),
colour_columns_by=c("cluster", "Plate", "Oncogene"),
cluster_cols=FALSE, center=TRUE, symmetric=TRUE, zlim=c(-5, 5))