写在前面,这篇文章主要整理自于Gene co-expression analysis for functional classification and gene–disease predictions,这是我翻译并进行注释的比较长的一篇文章了。
先把文章最后的key notes列出来:
Key Points
• RNA-seq-based co-expression analysis can be used to assign putative functions to non-coding RNAs and to identify candidates for roles in disease.
• In co-expression networks, hub gene identification has a limited power for identifying targets for follow-up studies; yet, this can be enhanced by integrated net-work analyses, which may incorporate GWAS hits, eQTLs, TFBSs and other data layers.
• Differential co-expression analyses can reveal genes that have different co-expression partners between healthy and disease state and can help to uncover regulators underlying disease and other phenotypes.
• Methods such as biclustering and Generalised Single Value Decomposition (GSVD) allow the identification of signals/modules unique to specific cancer subtypes, which may serve a purpose in prognosis and for preci-sion medicine.
经典的共表达网络构建和分析由以下3步构成
第一步,基于每对基因间的相关性或相互信息定义每2个基因之间的个体关系。这些关系描述了所有样本中基因对表达模式之间的相似性。不同的相关性方法已经被用了构建网络,包括皮尔逊或斯皮尔曼相关系数。可选择的,最小绝对误差回归或贝叶斯方法也可以用来构建共表达网络。后2个有个附加优势,因为他们可以用来鉴定随机联系,这已经在其他地方描述。对于其它相似性方法的讨论,请参考参考文献30。很多这种相似性矩阵也可以用了构建PPI网络,已经用癌症数据进行比较了,ref31。
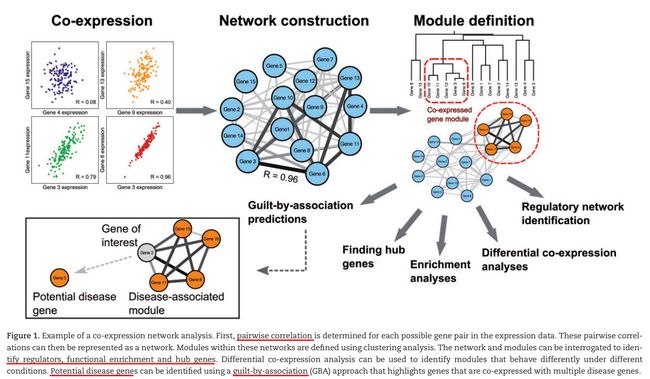
第二步,共表达相关性用了构建网络,每个node代表一个gene,每条边代表共表达关系的强度。下面fig1。
第三步,使用其中一种可行的聚类方法鉴定modules(共表达基因groups)。共表达分析的聚类用来把所有样本中相似genes归组,产生共表达基因group而不是仅仅共表达基因对。这种聚类方法在选择的时候需要考虑,因为这会严重影响结果,和分析的意义。许多聚类方法是可行的,包括k-means聚类和等级聚类,这在ref33中。Modules然后可以被功能富集分析解释,这是一种鉴定排序在一个genes list中overrepresented功能范畴的方法。
共表达分析中,考虑样本的异质性非常重要。组织特异性或condition特异性共表达网络模块在一个从多组织或多condition而来的共表达网络中不能被检测到,因为组织或condition特异性模块的相关性信息因为其他组织/condition缺乏相关信息被稀释了。然而,把共表达分析限制在一个特定的组织或condition也会减少样本size,因此会降低统计效应而不能检测到共表达模块。因此,不区分组织或condition间的方法应当用来鉴定普通共表达模块。而用来比较不同组织或condition的差异共表达更好的用来鉴定对特定的组织或condition特异的模块。
1 共表达网络的类型
Signed和unsigned共表达网络(有方向和无方向,起始是正相关和负相关)
在一个基于相关性的共表达网络中,相关性测量的值介于-1(完全负相关)到1(完全正相关)之间。在无方向网络中,使用的是绝对相关系数,这就以为着两个负相关的genes也会被认为共表达,这就导致负相关的genes也会被group到一起。因为另外那些完全正相关共表达的genes是完全不同的表达模式,这些正负相关genes被group到同一个模块中,可能会扰乱网络结构。有方向的网络可以解决这个问题,其采取的是把相关性值scale到0和1之间。其中,0-0.5代表负相关,0.5-1代表正相关。有方向的方法产生的网络可以更好的分离出有生物意义的modules(比如那些代表一个特别的生物过程的)。这样,接近0的值代表负相关,当miRNAs被引入网络中时,这个特征可能尤其有意义,因为miRNAs主要通过下调其他genes来行驶其功能。这对一些长基因间非编码RNAs(lincRNAS, long intergenic non-coding RNAs)同样生效。
Weighted and un-weighted co-expression networks
在一个权重网络中,所有的genes都彼此联系,这些联系的值介于0和1之间,以此来显示这些genes之间的共调控强度。在一个非权重网络中,基因对之间的关系是二进制,0或1,代表gene之间要么没联系要么有联系。一个非权重网络可以从权重网络产生。例如,考虑相关性大于某个值的genes有联系,否则没有。我们review关注权重网络,因为到目前位置,他们比非权重网络产生的网络更加稳健,ref40。
Microarrays versus RNA-seq data
从微阵列或RNA-seq技术获得的data都可以从他们的表达谱数据构建共表达网络。RNA-seq的一个最大的优势是它可以对超过7w个非编码RNAs的表达值进行定量,这对微阵列是不可能做到的,这其中包括最近刚被注释的lincRNAs,他们中的很多都被认为有调节作用,在疾病中要重要角色。因此,为了生物过程驱动的调控机制的理解,非编码RNAs需要考虑分析。
RNA-seq还有其他优势。它提高了低丰度转录本的精确性,对鉴定组织特性表达,区分紧密相关的旁系同源genes有更好的解决。RNA-seq也可以鉴定不同剪切体之间的表达,它们有不同的相互作用partners和生物功能。对RNA-seq的共表达分析可以这些剪切体和lincRNAs指定推断的功能,并且可以推断他们可能参与的疾病。剪切体水平的共表达分析的局限是引入了偏倚性,因为如果很多剪切体共享同一个表达的外显子的话,很难发现那个剪切体被表达。
作为RNA-seq’s的应用的一个例子是isoform和外显子特异表达水平测量,外显子水平表达用来构建co-splicing 网络。在基因共表达网络中,同一个genes来源的不同转录本的表达通常会被加强,这会导致有偏倚的共表达信号。在一个co-splicing网络中,这个问题可以在计算基因共表达系数时,通过考虑外显子表达水平一个gene内的分布来解决。在一个生物学terms这意味这,只有两个基因的不同的splice variants显示出共调节表达他们才被考虑相关,如果不是这样,那么他们被认为不是共表达,甚至这个基因的总体表达谱是相关的。这个方法已经鉴定了新的功能modules,这使用传统的共表达网络是无法鉴定的。另外,使用这种方法,含有很多个外显子和转录本的基因需要在网络中有更多的relevent位置。
一个不同的方法是基于reads匹配到不同外显子上分布来决定源于同一个gene的不同同分异构体的表达。这个方法是SpliceNet。
2 Clustering and network analysis identifying modules
聚类用来把在很多样本中有相似表达模式的genes进行归组,这些产生的模块经常代表某生理过程,或某特殊表型。
用的最多的共表达分析的聚类包是WGCNA。这个包使用的是等级聚类构建共表达网络,当然这个是基于表达谱数据的相关性。等级聚类,把每一个cluster分成sub-clusters来产生带树枝的树,这些树枝代表共表达模块。然后这些模块通过剪切树枝的高度来进行定义。
WGCNA是第一个用于RNA-seq数据共表达网络构建的工具,和微阵列数据方法相似。基于这个方法,鉴定了一个lincRNAs模块,这个模块和心脏病有关。
共表达模块还被用了鉴定人和小鼠不同的发育阶段。每个阶段的每个模块被鉴定出来然后在人和小鼠之间进行比较,揭示了在小鼠oocyte形成和人中oocyte和单细胞阶段的共表达模块的强overlap。这表明,人和小鼠在早期发育阶段有共同的核心转录程序,后来不一样了。Ref12。
Identifying modules
聚类用来把在很多样本中有相似表达模式的genes进行归组,这些产生的模块经常代表某生理过程,或某特殊表型。
用的最多的共表达分析的聚类包是WGCNA。这个包使用的是等级聚类构建共表达网络,当然这个是基于表达谱数据的相关性。等级聚类,把每一个cluster分成sub-clusters来产生带树枝的树,这些树枝代表共表达模块。然后这些模块通过剪切树枝的高度来进行定义。
WGCNA是第一个用于RNA-seq数据共表达网络构建的工具,和微阵列数据方法相似。基于这个方法,鉴定了一个lincRNAs模块,这个模块和心脏病有关。
共表达模块还被用了鉴定人和小鼠不同的发育阶段。每个阶段的每个模块被鉴定出来然后在人和小鼠之间进行比较,揭示了在小鼠oocyte形成和人中oocyte和单细胞阶段的共表达模块的强overlap。这表明,人和小鼠在早期发育阶段有共同的核心转录程序,后来不一样了。Ref12。
Identifying hub genes
通过clustering鉴定的共表达模块通常很大,这样的话,就非常有必要来鉴定每个模块中哪些基因可以更好的解释这种行为。一个广为使用的方法是鉴定共表达网络中的高连接度genes(highly connected genes),也就是hub genes。Hubs比起其他nodes来说,和网络的功能更加相关ref69,在生物网络中也是这样32,虽然数学派生词显示,这只是对于intra-modular hub gene模块内hub genes。(和inter-modular hub genes相反)64,65。模块内的hubs是网络中某特定模块中最重要的,支配作用的。为了鉴定hub genes,centrality 方法,主要是“betweenness centrality”常被使用。那些有高的中介性中心值的基因(high betweenness centrality)作为网络中最短路径连接(子),非常重要。连接度通常用来度量网络的稳健性robustness,显示了在保持基因不被连接之前有多少genes需要从网络中被移除。鉴定共表达网络中的hub genes已经协助发现cancer,II型糖尿病,和其他疾病中的必须基因。还有组织再生。
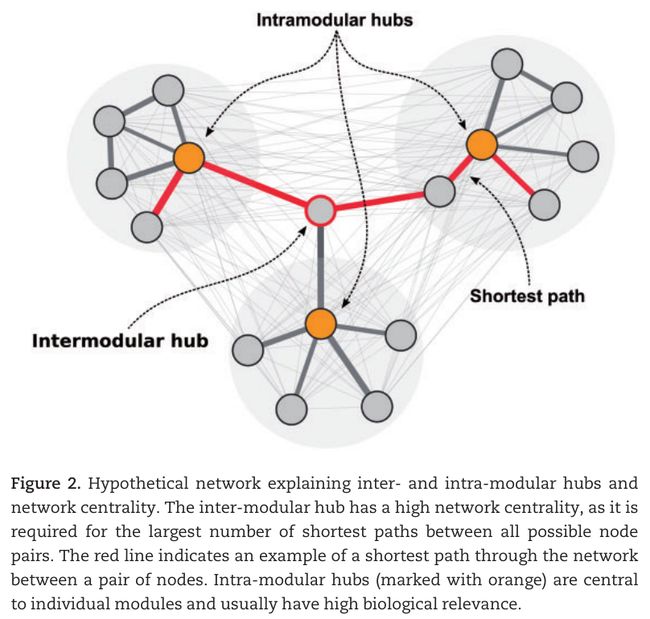
因为在一个module中通常有很多hubs或差异连接genes,通常对隐含在表型下最重要的gene并不是十分清晰。也没有保证就说hub genes就和表型有因果关系。使用来自1617个样品的心肌数据的一项研究发现已知的胎儿基因标志物在发育和患病心肌共同的基因网络中上调不是枢纽基因。另一个有关沙门菌的共表达研究发现,hub genes对生长,压力调节,毒力等可有可无,这都揭示,hub genes不是一定必不可少。(头大了。。。。。。。。。。。。。)
Guilt by assosiation
把生物意义附加到modules中,一个广为使用的方法是确定一个模块内的genes的功能富集,这可以使用下面这个table1中的工具。(table1很长,覆盖了本文中所有流程工具)。
假定共表达基因是功能相关的,假设共表达的基因在功能上是相关的,可以将富集的功能分配给相同共表达模块内的注释不佳的基因,这种方法通常被称为“通过关联犯罪”(GBA)121(类似株连,大家都在一个模块内,别的都有这种功能,那你虽然没被注释,但很可能也这样)。GBA方法也被广泛用于鉴定新的潜在的疾病基因,如果一个模块内的genes的大部分都和一个特定的疾病联系在一起的话,26,121-126,fig1。
当使用GBA方法时,一定要记住,并不是模块中的每个基因都一定与它所富集的功能或疾病相关。因为共表达模块通常由大量的genes组成,任何功能过程或疾病相关的基因group的过量表达都会迅速变的有统计学意义,切p值很小。对这些p值的误解可能会导致一个错误的结论,那就是一个模块内的所有genes都在一个特定的过程或疾病中起着重要作用。而实际上,模块中与其主要生物功能相关的genes比例通常都小于20%,参考127,并且,module-trait相关性可能相对较低(相关性<0.5),哪怕有统计学意义。128。(28法则么,每段手法一致,先说好,再说不好,再引入下一个section)
Regulatory network construction
虽然有足够的证据表明,共表达网络分析可以协助鉴定在疾病或生物功能中起重要作用的基因,但是从共表达网络中推论因果关系仍然很难。像ARACNE(23)和GENIE3(113)这样的工具试图从共表达网络总构建调控网络。ARACNE移除genes间的非直接连接(例如,与基因本身相关性较强的基因的伙伴),只留下可能有调节作用的连接。GENIE3整合了TF信息来构建调控网络,这通过最能解释每个靶基因表达模式的TF的表达模式。GENIE3的局限是,A limitation of GENIE3 is that TF information is required for it to perform better than random chance。
(后面还有很多,大概就是wgcna结合ARACNE可以做的很好)。131
Differential co-expression analysis
差异共表达分析可以鉴定生物学上重要的差异共表达模块,这使用常规的共表达或差异表达分析是不能的。那些不同的样本groups中差异共表达的基因更可能是regulators,因此更能解释表型之间的差异。差异共表达分析已经被用来鉴定隐含在健康和疾病样本间或不同组织,细胞或种族间的genes。下面我们提供通常使用的和新出现的方法和工具,可以分为两个范畴。第一,鉴定预先定义的样本组之间的差异共表达的方法(比如conditions,时间点或组织类型)第二,不需要关于样本组的先验知识,使用的是一个算法来鉴定共表达clusters(预先不知道样本subpopulations)。
Differential co-expression analysis between sample groups
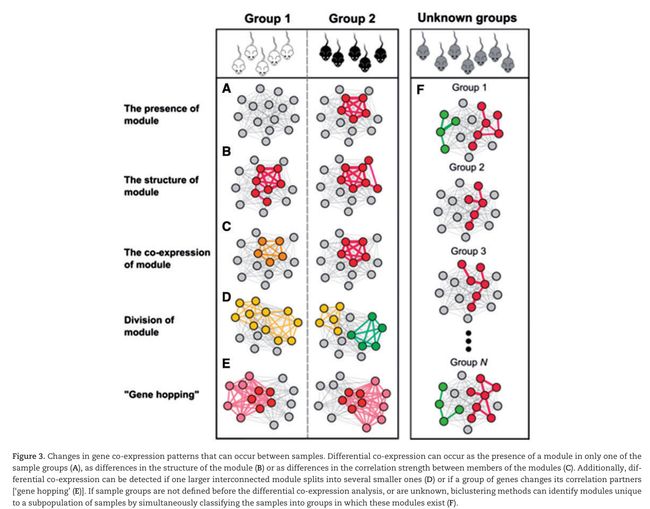
大多数的差异共表达分析依赖不同clustering,他们鉴定的是clusters,这些clusters包含的是在改变的conditions或表型下不同的基因或行为异同。最常使用的进行差异聚类分析的程序(也已经和其他程序比较)是WGCNA.,DICER,DiffCoEx,所有以上这些都是先鉴定所有研究样本中共表达的模块。这些共表达模块然后可以和预先定义的样本亚群关联,比如,疾病状态或组织类型。
WGCNA定义每个样本亚群中module的活性和重要性fig3a3c。对每一个module,会计算一个特征基因,这个向量可以最好的描述这个模块中所有基因的表达行为。然后,通过鉴定与模块基因组行为相似的基因或模块内基因的基因(这些基因往往一致),优先考虑这些模块中的哪些基因可能成为与模块相关的表型的基础。(以下关于那两个方法的不再翻译)
下面作者讲了一些应用
比如,组织特异性网络中,对组织功能特异的TFs倾向于和组织特异性基因一起高表达。这些基因之间相对其他基因更倾向于形成强连接,这些基因倾向于与其他基因形成更强的连接,但是保留在网络的外围(因此具有低的中心性),而组织特异性的TF变得更加重要。因此,通过识别组织特异性网络(图3A和3C)中增加的共表达强度的模块以及通过精确定位这些模块的中心枢纽,可以揭示组织特异性TF。相反,通过鉴定这些模块周围的基因,可以检测到不是TFs但是组织特异性的基因(图3B)。 而且,一些TF在不同的组织中具有不同的作用。 这些TF将被认为是在一个条件下是一个模块核心的中枢基因,而在另一个条件下是另一个模块的核心。差异连接的基因是在两个样品组之间具有不同共表达伴侣的基因。 这些基因似乎在两组间观察到的表型差异中发挥调节作用(图3D)例如,一项研究使用类似于DiffCoEx的方法比较了突变牛中的共表达与增加的肌肉生长与非突变体中的共表达。 通过鉴定最差异表达的基因和显示与这些基因的最高差异连接的TF(图3D)(图3D),鉴定了含有因果突变(肌肉生长抑制素)的TF。 有趣的是,编码这种TF的Mstn基因在表达本身中几乎没有变化,提供了差异共表达分析如何揭示生物学上重要的发现而不是仅通过差异表达分析揭示的例子。
3 Integrated network analysis
实验验证经常是聚焦于单个基因。因为这些实验花费多又耗时,所以对因果基因的高可信度预测非常重要。仅仅基于共表达的分析还不能提供这种信心。因此,整合其他类型的数据有助于优先考虑那些可能构成表型的genes。这是可行的,例如,使用诸如描述那些基因是TF是,这可以由GENIE3进行调控预测。然而,聚焦于TFs是不够的,还需要整合多个数据类型来提高网络的精确性和有效性。
TF binding site analysis
基因组范围的转录因子结合位点(TFBS)分析是在本世纪初采用的是染色质免疫沉淀,然后是芯片分析,也称为ChIP芯片,后来被替换为更精确的ChIP-seq [149]。这些数据被用来从基因表达和TFBS数据中创建全基因组整合调控网络[150]。 基于ChIP芯片的TFBSs和表达数据的联合分析初步显示,在58%的case中,与基因启动子区域结合的TFs确实被相应的TF调控[151]。 偏最小二乘法(一种众所周知的分析方法高维数据与几个连续响应变量)后来被提出来识别假阳性,并区分TFs的激活和抑制活性[152]。 更新的方法利用快速增加的ChIP-seq数据的可用性,结合表达数据对TF结合的基因进行排序,这可以用来优先选择最可能的TF目标[153]。 进行类似分析,整合表达和ChIP数据的工具也已经出版。
Multilayer integrated networks
独立于识别他们的方法,共享eQTL基因靶标,TF/miRNA靶标或富集motifs的网络模块可以被进一步研究。几种计算方法和公用可获得数据集可以用了进行多组学数据整合。例如,关于eQTLs的信息可以从最近的large-scale blood-based trans-eQTL meta分析获得,或者从其他组织类型进行的Eqtl研究获得。转录因子结合位点TFBSs可以从JASPAR和DeepBind数据库获取,这两个网站包含从实验数据来的TF结合motifs。通过investigate查看从ENCODE来的组织特异性CHIP-seq 峰,结合位点可以被进一步优先化。最后,使用几个生物信息学靶基因预测工具158,159可以鉴定miRNA-target的相互作用,或者使用手工产生的实验支持的数据库来探索这些靶基因相互作用160-162。
整合不同层次的数据信息或许会以几种方式产生新的生物学可解释的联系。如果模块内的hub genes是TFs或TF的靶标,那么这个TF很可能和你研究的表型具有因果作用。如果在同一模块中存在多个全基因组关联研究(GWAS)命中,则其累积存在可以显着促成疾病发展。120.163.164。在一个共表达模块中的gene的不同甲基化状态也可以阐明潜在的疾病的模式。如果多个基因受到相同遗传变异的调控(在trans-eQTL效应下),通过鉴定驱动trans-eQTL效应的cis-eQTL基因,可能可能鉴定出负责改变网络的基因(图4)。这个是被下面这个事实支持的:这得到以下事实的支持:疾病相关基因组变体的反式调节下的基因有时在功能上与相应疾病相关的过程或途径相关。
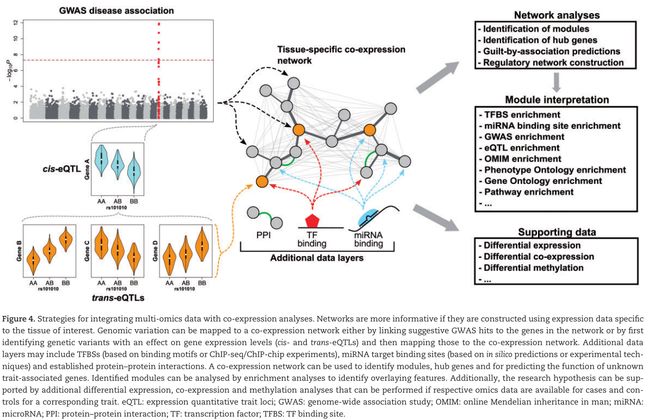
总之,整个多个数据类型增加结果预测的精确性。例如,通过将肿瘤基因组序列与基因网络整合,鉴定了不同亚型癌症所特有的模块[166],这些模块可能有助于个体化医疗治疗推断的靶标的预后和鉴定。 本文前面介绍的一些工具可用于差异共表达分析,但也可应用于其他数据类型。 在最初的DINGO出版物中,作者对mRNA表达,DNA拷贝数变异和甲基化数据进行了综合分析。通过覆盖每种数据类型的差异网络,识别出他们所有中都出现的边,来自PI3K通路的很多genes被识别为恶性胶质瘤病人的重要players。这个通路是一个已经建立的治疗靶点,支持这个notion:在疾病相关靶标的识别上是一个有效的方法。一个最近刚出版的tool,CoRegNet,通过识别不同数据类型的基因共合作调节(co-operative regulators of genes)允许不同类型数据的整合到一个共表达分析中。另外一个已经建立的方法是cMonkey,通过识别多个数据类型中的可group到一组的gene组来计算联合双聚类成员的可能性,以此获取相似的数据整合。
4 Future prospects
近些年来,差异共表达分析更多的被用来分析大的数据集。这归因于:大规模基因表达谱花费降低,尤其是RNA-seq;增加的样本sizes;从扰动实验来的组织特异性数据的更大的可获取性,这些都是硕果累累的差异共表达分析的原因。
另外一个前景是来于RNA-seq数据的突变检测。因为在不同的细胞中突变会随着年龄累积,这可以用于发现cell的来源。突变累积已经用来研究癌症发展和恶变来源。在大规模单细胞RNA-seq实验中,可以基于其来源进行分离细胞,或者基于他们拥有的突变来group细胞。拥有相同的突变的细胞可以发现有共表达模式,带有特异突变的细胞模块也可以被检测。这可以直接把图表和表达模块联系起来,有个局限就是在RNA-seq中只有编码区的突变区域才能被识别。随着不同类型数据诸如RNA-seq,基因组序列,CHIP-seq,甲基化和蛋白组数据的持续增长,整合这些数据来精确预测调控基因成为可能。来自像GTEx,Epigenome Roadmap,ENCODE这种大集团的项目,已经产生了来于多个组学水平的数据,促进了整合分析。为了识别调控关系,扰动数据是更合适的,因为cononical数据不能区分调控关系中的真阳性和假阳性。进一步,调控关系可以是高度细胞类型-,组织-,或发育阶段-特异性。当前只有少量的工具和方法可以用来研究investigate多组学数据,并且现有的工具大部分只能整合两个层次的组学数据。整合网络分析有另外的数学挑战,最好的实践远没有构建。关于这些topic的进一步研究,对研究者十分有趣,因为这会对可以解释共表达模式和疾病机制的调控机制有更好的理解。而对这些疾病机制和相应的共表达模式的更好的理解便于确定干预研究的适当目标。