
HOMER 是一套用于Motif查找和二代数据分析的工具。HOMER 中的工具是使用Perl 和C++编写的,是Linux command line based。HOMER 这个软件是一个大杂烩,能解决几乎所有的高通量测序数据的分析。
1.HOMER软件安装和配置
homer软件的安装
使用conda安装,一句话搞定:conda install -c bioconda homer 。
使用configureHomer.pl完成Homer软件的配置
wget http://homer.salk.edu/homer/configureHomer.pl #首先下载configureHomer.pl
perl configureHomer.pl -install #使用configureHomer.pl配置Homer
configureHomer.pl配置Homer的语法是:(下一节讲具体应用)
perl /path-to-homer/configureHomer.pl [options]
我们看下这些options都有哪些:
Options:
-list 列出可用的包
-install 安装homer需要用到的数据包
-version 安装homer包时,可以指定包版本
-remove 移除包
-update 更新所有包到最新版本
-reinstall 强制重新安装所有已经安装过的包
-all 安装所有包
-getFacts (add humor to HOMER - to remove delete contents of data/misc/)
-check 检查第三方软件:samtools, DESeq2, edgeR
-make 重新配置和编译可执行文件
-sun SunOS系统,使用gmake 和 gtar代替make 和 tar
-keepScript 不更新configureHomer.pl
-url 安装时,使用的资源地址,默认:http://homer.ucsd.edu/homer/
Hubs & BigWig settings (with read existing settings from config.txt if upgrading):
-bigWigUrl(Setting for makeBigWigs.pl)
-bigWigDir(Setting for makeBigWigs.pl)
-hubsUrl(Setting for makeMultiWigHub.pl)
-hubsDir(Setting for makeMultiWigHub.pl)
Configuration files: 下载 update.txt,更新config.txt
2. HOMER包介绍和安装
homer包分为4种:
- SOFTWARE:homer工具包,包含一些常用数据。
- ORGANISMS:物种特异性的数据,包含Gene accession, gene descriptions, and GO analysis信息。大多数数据来自于NCBI Gene database。下载promoter 或 genome 数据时,会自动下载对应Organism 包。
- PROMOTERS:Promoter 序列信息和Promoter 富集分析的文件。大多数数据来自RefSeq transcript。
- GENOMES:基因组序列及其注释信息。
这里需要特别注意下:虽然conda安装好了Homer,但并没有包含参考序列或注释数据 。所以需要使用 configureHomer.pl下载各种数据包
下载数据之前先查看数据列表,看Homer已经有哪些数据包:
语法公式:perl /path-to-homer/configureHomer.pl -list
具体事例:perl /home/jhuang/miniconda2/share/homer-4.9.1-6/configureHomer.pl -list
等同于:perl /home/jhuang/miniconda2/share/homer-4.9.1-6/.//configureHomer.pl -list
#主要是给系统一个configureHomer.pl路径
如上述介绍,Homer数据包安装使用-install参数
perl /path-to-homer/configureHomer.pl -install mouse #下载老鼠启动子数据
perl /path-to-homer/configureHomer.pl -install mm10 #下载 mm10小鼠参考基因组
perl /path-to-homer/configureHomer.pl -install hg19 #下载 hg19人的参考基因组
具体实例:
nohup perl /home/jhuang/miniconda2/share/homer-4.9.1-6/.//configureHomer.pl -install hg19 &
#太慢了所以挂后台了
3.利用ChIP-Seq结果在基因组区域中寻找富集的Motifs
首先对bed文件进行处理
用MACS2软件进行peak calling,也就是找比对结果(bam文件)的peaks,MACS2找到的peaks会存放在生成的*.bed文件里。homer软件找motif需要读取我们这里得到的bed格式的peaks文件。homer软件不仅可以找到motif,还可以注释peaks。
homer软件在读取bed文件时,需要提取对应的列作为输入文件
我们要对MACS找到的peaks记录文件,还需提取对应的列给HOMER作为输入文件,提取操作为:
awk '{print $4"\t"$1"\t"$2"\t"$3"\t+"}' sample_peaks.bed >sample_homer.bed
不理解没关系,我们拿我跑的chipseq数据做示范,看awk对bed文件做了什么
使用命令less -S ChIP-Seq_H3K4Me3_1_trimmed_rmdup_summits.bed 看下awk处理之前bed文件长什么样子:
$ less -S ChIP-Seq_H3K4Me3_1_trimmed_rmdup_summits.bed
chr1 29202 29203 ChIP-Seq_H3K4Me3_1_trimmed_rmdup_peak_1 260.18610
chr1 714432 714433 ChIP-Seq_H3K4Me3_1_trimmed_rmdup_peak_2 165.19276
chr1 762743 762744 ChIP-Seq_H3K4Me3_1_trimmed_rmdup_peak_3 195.10117
chr1 839761 839762 ChIP-Seq_H3K4Me3_1_trimmed_rmdup_peak_4 36.37314
chr1 859839 859840 ChIP-Seq_H3K4Me3_1_trimmed_rmdup_peak_5 122.88363
chr1 876411 876412 ChIP-Seq_H3K4Me3_1_trimmed_rmdup_peak_6 20.77421
chr1 877742 877743 ChIP-Seq_H3K4Me3_1_trimmed_rmdup_peak_7 77.55285
chr1 894490 894491 ChIP-Seq_H3K4Me3_1_trimmed_rmdup_peak_8 256.37842
chr1 896069 896070 ChIP-Seq_H3K4Me3_1_trimmed_rmdup_peak_9 183.38295
使用awk对bed文件进行处理,处理完的文件命名为ChIP-Seq_H3K4Me3_1_homer.bed
awk '{print $4"\t"$1"\t"$2"\t"$3"\t+"}' ChIP-Seq_H3K4Me3_1_trimmed_rmdup_summits.bed >ChIP-Seq_H3K4Me3_1_homer.bed
$ less -S ChIP-Seq_H3K4Me3_1_homer.bed
ChIP-Seq_H3K4Me3_1_trimmed_rmdup_peak_1 chr1 29202 29203 +
ChIP-Seq_H3K4Me3_1_trimmed_rmdup_peak_2 chr1 714432 714433 +
ChIP-Seq_H3K4Me3_1_trimmed_rmdup_peak_3 chr1 762743 762744 +
ChIP-Seq_H3K4Me3_1_trimmed_rmdup_peak_4 chr1 839761 839762 +
ChIP-Seq_H3K4Me3_1_trimmed_rmdup_peak_5 chr1 859839 859840 +
ChIP-Seq_H3K4Me3_1_trimmed_rmdup_peak_6 chr1 876411 876412 +
ChIP-Seq_H3K4Me3_1_trimmed_rmdup_peak_7 chr1 877742 877743 +
ChIP-Seq_H3K4Me3_1_trimmed_rmdup_peak_8 chr1 894490 894491 +
ChIP-Seq_H3K4Me3_1_trimmed_rmdup_peak_9 chr1 896069 896070 +
把bed文件变成这个样子,用于Homer软件的读取,我们称这样的文件叫做 Homer Peak/Positions file
我们可以看到Homer Peak/Positions file有4列:
- 第一列: 染色体
- 第二列: 起始位置
- 第三列: 终止位置
- 第四列: 链的方向(+/- or 0/1, where 0="+", 1="-")
寻找富集Motifs
findMotifsGenome.pl命令用于在基因组区域中寻找富集Motifs
命令使用格式:
findMotifsGenome.pl 使用实例:寻找我们刚刚生成的Homer Peak/Positions file文件:ChIP-Seq_H3K4Me3_1_homer.bed的 motif 使用命令:
findMotifsGenome.pl ChIP-Seq_H3K4Me3_1_homer.bed hg19 ChIP-Seq_H3K4Me3_1_motifDir -len 8,10,12
# 参数解释
-输入文件:awk处理好的Homer Peak/Positions file
-参考基因组:这里是hg19
-输出文件:给一个路径和输出文件的名字
-len:motif大小设置,默认8,10,12;越大需要的计算资源越多
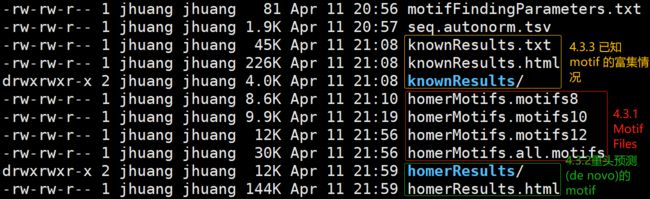
上述命令(找motif)每一样品需要运行30-40分钟后,得到文件夹ChIP-Seq_H3K4Me3_1_motifDir,文件夹里的内容有:
-rw-rw-r-- 1 jhuang jhuang 30K Apr 11 21:56 homerMotifs.all.motifs
-rw-rw-r-- 1 jhuang jhuang 9.9K Apr 11 21:19 homerMotifs.motifs10
-rw-rw-r-- 1 jhuang jhuang 12K Apr 11 21:56 homerMotifs.motifs12
-rw-rw-r-- 1 jhuang jhuang 8.6K Apr 11 21:10 homerMotifs.motifs8
drwxrwxr-x 2 jhuang jhuang 12K Apr 11 21:59 homerResults
-rw-rw-r-- 1 jhuang jhuang 144K Apr 11 21:59 homerResults.html
drwxrwxr-x 2 jhuang jhuang 4.0K Apr 11 21:08 knownResults
-rw-rw-r-- 1 jhuang jhuang 226K Apr 11 21:08 knownResults.html
-rw-rw-r-- 1 jhuang jhuang 45K Apr 11 21:08 knownResults.txt
-rw-rw-r-- 1 jhuang jhuang 81 Apr 11 20:56 motifFindingParameters.txt
-rw-rw-r-- 1 jhuang jhuang 1.9K Apr 11 20:57 seq.autonorm.tsv
其中会生成一个详细版的网页结果,生成这些文件的具体作用下节讲述
常用参数:
-bg:自定义背景序列;
-size: 用于motif寻找得片段大小,默认200bp;-size given 设置片段大小为目标序列长度;越大需要得计算资源越多;
-len:motif大小设置,默认8,10,12;越大需要得计算资源越多;
-S:结果输出多少motifs, 默认25;
-mis:motif错配碱基数,默认2bp;
-norevopp:不进行反义链搜索motif;
-nomotif:关闭重投预测motif;
-rna: 输出RNA motif,使用RNA motif数据库;
-h:使用超几何检验代替二项式分布;
-N:用于motif寻找得背景序列数目,default=max(50k, 2x input);耗内存参数
使用 annotatePeaks.pl 对peaks进行注释
命令使用格式:
annotatePeaks.pl 1>output.peakAnn.xls 2>output.annLog.txt
使用实例:注释ChIP-Seq_H3K4Me3_1_homer.bed的 peaks 使用命令:
annotatePeaks.pl ChIP-Seq_H3K4Me3_1_homer.bed hg19 1>ChIP-Seq_H3K4Me3_1_peakAnn.xls 2>$ChIP-Seq_H3K4Me3_1_annLog.txt
4.HOMER Motif 分析基本步骤和结果分析
Homer主要被用于 ChIP-Seq 分析,但是核酸序列motif寻找问题都可以尝试使用。
注意:这里大部分是在介绍Homer软件背后是怎么工作的,不是说我们在使用Homer软件找motif时的步骤,我们使用的时候直接调用Homer软件的内部命令就好
4.1 预处理
一、提取序列
提供的数据是基因组位置信息,就需要提取对应的DNA信息;提供基因号时,需要选择启动子区域。对应着就是我们前面用awk 处理bed文件,最后得到要求的那四列。
二、背景选择
未指定背景序列时,HOMER 会自动选择。(上面chipseq处理的时候就没有指定背景序列)
对基因组某些区域进行分析时,从基因组随机选择GC含量一致的序列作为背景序列。
对启动子进行分析时,除用来分析外的所有启动子将被作为背景。
自定义背景使用参数-bg
三、GC 标准化
目标序列(对应着上面的就是Homer Peak/Positions file)和背景序列会基于GC含量按5%作为bin 查看GC含量的分布。背景序列会得到权值,从而使得其GC含量分布与目标序列一致。
ChIP-Seq 实验得到序列GC含量。
四、自动标准化
需要分析的序列除了GC含量会带来误差,其他的生物学现象,外显子中密码子偏好性或测序实验中偏好性都会影响分析。对于足够强的偏差,HOMER 会自动追踪目标序列和背景中显著差异的特征序列,并通过调整背景序列的权重来平衡输入数据和背景中短寡聚核酸序列不平衡。
短寡聚核酸序列长度可以通过参数-nlen <#>指定。
4.2 重头预测Motifs
默认情况下,HOMER 调用homer2 进行motif 分析;通过参数"-homer1" 可以指定老版本工具。
一、将输入序列解析为寡聚核苷酸序列
将输入序列按照motif 长度期望值解析为寡聚核苷酸序列,以及创建Oligo 数据表。Oligo 数据表中记录着每条oligo 在目标序列和背景中被发现的次数。
二、Oligo 自动标准化
三、 全局搜索阶段
Oligo 表格信息构建好之后,HOMER 对富集的Oligo 进行全局搜索。如果一个Motif是富集的,那么属于这个Motif的Oligo 也应该会富集。首先,HOMER 会搜索可能富集的Oligo 。HOMER 允许错配 。
使用参数-mis <#> 调节允许的错配数目
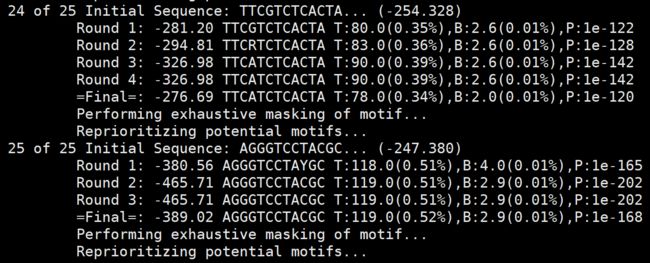
四、Mask and Repeat
当最优oligo被优化成motif后,motif 对应的序列从要分析的数据中移除,接下来再分析最优的.....直到 25(默认值,"-S <#>")个motifs 被发现。
比如,我们这里处理chipseq时的情况
五、计算已知Motifs是否富集
导入Motif库
为了搜索输入数据中已知Motifs ,HOMER 可以输入已知Motifs 数据,可以使用HOMER 默认的 ("data/knownTFs/known.motifs"),也可以是自己构建("-mknown
筛选每一个Motif
对于每个motif,HOMER 计算丰度(包含motif的序列/background sequences), ZOOPS (zero or one occurence per sequence)计数以及使用超几何检验或二项式计算显著性。
4.3 Motif 的结果分析
我们chipseq,找motif的输出文件如下图:
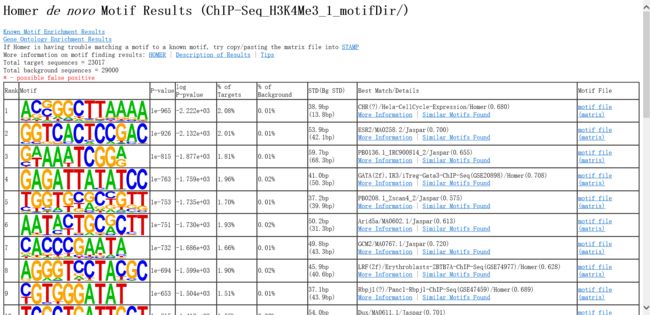
这些输出结果包括:
4.3.1 Motif Files
*.motif 文件包含motifs的信息
"*.motif"文件格式:
>ATATTGCG 1-ATATTGCG 10.055970 -947.021323 0 T:434.0(1.88%),B:22.3(0.08%),P:1
0.997 0.001 0.001 0.001
0.001 0.001 0.001 0.997
0.997 0.001 0.001 0.001
0.001 0.363 0.001 0.635
0.001 0.001 0.001 0.997
0.001 0.001 0.997 0.001
0.001 0.997 0.001 0.001
0.001 0.001 0.997 0.001
>AAAATCGG 2-AAAATCGG 10.633913 -833.891683 0 T:458.0(1.99%),B:34.6(0.13%),P:1
0.997 0.001 0.001 0.001
0.997 0.001 0.001 0.001
0.997 0.001 0.001 0.001
0.997 0.001 0.001 0.001
0.351 0.001 0.001 0.647
0.001 0.997 0.001 0.001
0.001 0.001 0.997 0.001
0.001 0.001 0.997 0.001
一个motif 的信息分为一块。motif 信息首行是motif 各种统计信息;其他行对应各个A/C/G/T的占比。
motif 信息首行解析:
- ">" + 序列 (可能是空白) example: >ASTTCCTCTT
- Motif 名字 example: 1-ASTTCCTCTT or NFkB
- 检测阈值对数值 example: 8.059752
- 富集P-value对数值 example: -23791.535714
- 0 用于老版本格式的占位符
- T:17311.0(44.36%),B:2181.5(5.80%),P:1e-10317
- T:#(%) - 包含motif的目标数据序列数除以目标数据序列总数
- B:#(%) - 包含motif的背景序列数除以背景序列总数
- P:# - 富集 p-value
- 用逗号分隔Motif统计量, example: Tpos:100.7,Tstd:32.6,Bpos:100.1,Bstd:64.6,StrandBias:0.0,Multiplicity:1.13
- Tpos: motif在目标序列中的平均位置 (0 = start of sequences)
- Tstd: motif在目标序列中位置的标准偏差
- Bpos: motif在背景序列中的平均位置 (0 = start of sequences)
- Bstd: motif在背景序列中位置的标准偏差
- StrandBias: log ratio of + strand occurrences to - strand occurrences.
- Multiplicity: 具有一个或多个结合位点的序列中每个序列的平均出现次数
4.3.2重头预测(de novo)的 motif
首先会对motif进行去冗余,将每个motif 的概率矩阵转换为向量,求motif之间的Pearson 相关性。
HTML 结果:
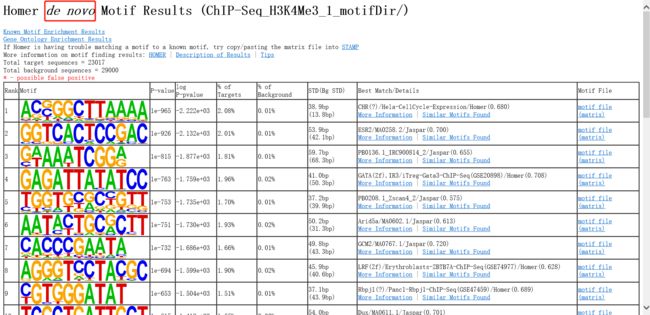
表格中,Best Match/Details项中:
More Information:与预测的motif相似的的已知motifs
Similar Motifs Found:与预测的motif相似的的其它预测motifs
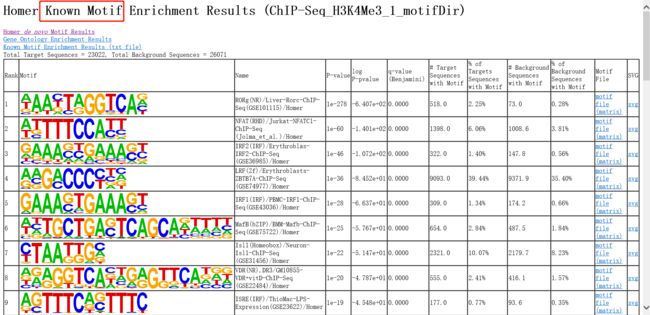
4.3.3 已知 motif 的富集情况(knownResults.html)
参考资料:
1.http://homer.ucsd.edu/homer/ngs/peakMotifs.html
2.https://www.jianshu.com/p/1384173c353b
3.https://www.jianshu.com/p/4af579b2e532
4.https://www.jianshu.com/p/2bf020cc8f90
5.https://www.jianshu.com/p/1602c2621c2b
6.https://www.jianshu.com/p/1602c2621c2b