
image.png

image.png

image.png

image.png

image.png

image.png
-
p 越小,效力越大
 image.png
image.png
 image.png
image.png
 image.png
image.png
 image.png
image.png
 image.png
image.png
 image.png
image.png
 image.png
image.png
 image.png
image.png
 image.png
image.png
 image.png
image.png
 image.png
image.png
 image.png
image.png
 image.png
image.png
 image.png
image.png
 image.png
image.png
 image.png
image.png
 image.png
image.png
 image.png
image.png
 image.png
image.png
 image.png
image.png
 image.png
image.png
 image.png
image.png
 image.png
image.png
 image.png
image.png
 image.png
image.png
 image.png
image.png
 image.png
image.png
 image.png
image.png
 image.png
image.png
 image.png
image.png
 image.png
image.png
 image.png
image.png
 image.png
image.png
 image.png
image.png
 image.png
image.png
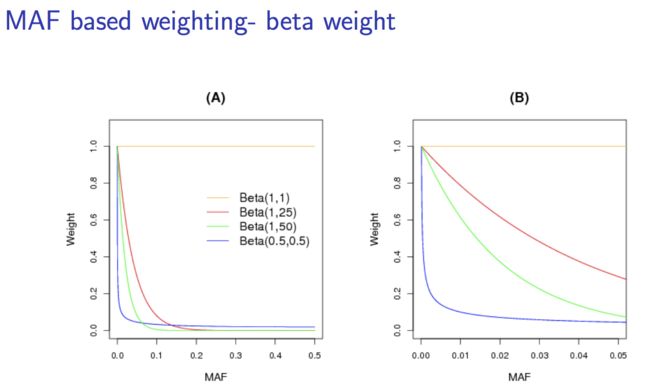 image.png
image.png
 image.png
image.png
 image.png
image.png
 image.png
image.png
##################################################################################################################
### R Scripts: let's read Gene2 into R and analyze it
library(SKAT)
Z = as.matrix(read.table("Gene2.txt", header = F))
maf = apply(Z,2,mean)/2 ## calculate the mean of each column and divide by 2 to get the maf
y.c = scan("Trait2.txt") ## read in the values for trait 1
rvs = which(maf<0.03) ## Set the threshold to be 0.03
obj<-SKAT_Null_Model(y.c ~ 1, out_type="C") ## calculates the NULL model, i.e. there are no variants
SKAT(Z, obj)$p.value ## test for trait 2 with gene 2. Note that we are using the default settings and testing all variants.
SKAT(Z[,rvs], obj)$p.value ## restrict attention to rare variants. The null model has not changed.
- 定性性状
##################################################################################################################
##################################################################################################################
### R Scripts: let's dichtomize trait 2
q1 = quantile(y.c,0.25)
q3 = quantile(y.c,0.75)
y.b = rep(NA, length(y.c))
y.b[which(y.c <= q1)] = 0
y.b[which(y.c >= q3)] = 1
omits = which(is.na(y.b))
y.b = y.b[-omits]
Z.b = Z[-omits,]
obj<-SKAT_Null_Model(y.b ~ 1, out_type="D") ## calculates the NULL model, i.e. there are no variants
SKAT(Z.b, obj)$p.value ## test for trait 2 with gene 2. Note that we are using the default settings and testing all variants.
SKAT(Z.b[,rvs], obj)$p.value ## restrict attention to rare variants. The null model has not changed.
-
gene2.txt 1000个人,62个位点
 image.png
image.png
##################################################################################################################
##################################################################################################################
### R Scripts: let's try running the C-alpha; recall that this can be done by setting the weights to be 1!
obj<-SKAT_Null_Model(y.c ~ 1, out_type="C") ## calculates the NULL model, i.e. there are no variants -- need to rerun because outcome changed
SKAT(Z[,rvs], obj, weights.beta = c(1,1))$p.value ## restrict attention to rare variants. The null model has not changed.
##################################################################################################################
##################################################################################################################
### R Scripts: omnibus version of SKAT with several different rho values
SKAT(Z[,rvs], obj, r.corr=0)$p.value ## same as running SKAT
SKAT(Z[,rvs], obj, r.corr=1)$p.value ## same as weighted count collapsing
SKAT(Z[,rvs], obj, r.corr=.5)$p.value ## something in between
weights = dbeta(maf[rvs], 1,25)
C = Z[,rvs]%*%weights ## calculate the collapsed variable
summary(lm(y.c~C))
##################################################################################################################
##################################################################################################################
### R Scripts: omnibus version of SKAT with "optimal" rho value
SKAT(Z[,rvs], obj, method="optimal")$p.value
SKAT(Z[,rvs], obj, method="optimal.adj")$p.value ## slightly better type I error control in the tails.
##################################################################################################################
##################################################################################################################
### R Scripts: read in PLINK example data and analyze the 10 genes there.
## define the file names and paths, note that the SSD and SSD.info don't exist yet
File.Bed<-"Example1.bed"
File.Bim<-"Example1.bim"
File.Fam<-"Example1.fam"
File.SetID<-"Example1.SetID"
File.SSD<-"Example1.SSD"
File.Info<-"Example1.SSD.info"
## To use binary ped files, you have to generate SSD file first.
## If you already have a SSD file, you do not need to call this
Generate_SSD_SetID(File.Bed, File.Bim, File.Fam, File.SetID, File.SSD, File.Info)
## Now we can open the SSD files and also run SKAT
FAM<-Read_Plink_FAM(File.Fam, Is.binary=FALSE)
y<-FAM$Phenotype
## To use a SSD file, please open it first.
## After finishing using it, you must close it.
SSD.INFO<-Open_SSD(File.SSD, File.Info)
## Number of samples
SSD.INFO$nSample
## Number of Sets
SSD.INFO$nSets
obj<-SKAT_Null_Model(y ~ 1, out_type="C")
out<-SKAT.SSD.All(SSD.INFO, obj)
out