参考自生信技能树:https://github.com/shenmengyuan/RNA_seq_Biotrainee
2个样品2个重复(2*2)
1_1
1_2
2_1
2_2
实用小工具
SAM flags查询(sam格式文件)

准备工作


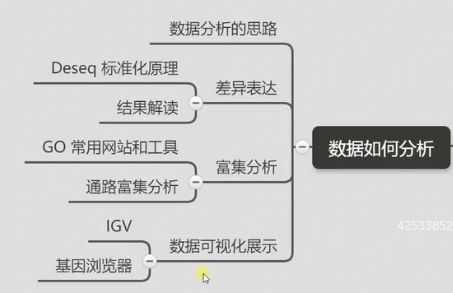
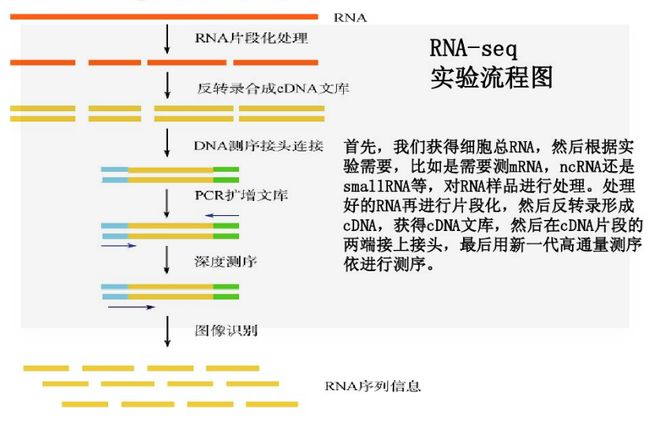
RNA-seq流程原理
1.准备工作
Using of software
FastQC Multiqc Trimmomatic STAR RSEM Subread HTSeq kallisto Deseq2 ...(conda可以安装)
查看软件是否可用,及时更新;准备好文件;
~/.bashrc
screen -S han #创建新窗口
# 会话为dead状态。使用screen -wipe命令清除该会话
# 鉴定是双端还是单端:计算fq.gz文件的reads;两个gz文件的reads数目相等,为双端测序;
zcat -f gKO-01.fq.gz |wc -l
# 参考基因组和注释下载;(示例而已)
[ftp://ftp.ensembl.org/pub/release-95/](ftp://ftp.ensembl.org/pub/release-95/)
# Genome file
[ftp://ftp.ensembl.org/pub/release-95/fasta/mus_musculus/dna/](ftp://ftp.ensembl.org/pub/release-95/fasta/mus_musculus/dna/)[Mus_musculus.GRCm38.dna.toplevel.fa.gz](ftp://ftp.ensembl.org/pub/release-95/fasta/mus_musculus/dna/Mus_musculus.GRCm38.dna.toplevel.fa.gz)
# GTF file
[ftp://ftp.ensembl.org/pub/release-95/gtf/mus_musculus/](ftp://ftp.ensembl.org/pub/release-95/gtf/mus_musculus/)[Mus_musculus.GRCm38.95.gtf.gz](ftp://ftp.ensembl.org/pub/release-95/gtf/mus_musculus/Mus_musculus.GRCm38.95.gtf.gz)
# 解压参考基因组和注释文件信息
gunzip *.gz
# 解压缩命令
tar -xzvf file.tar.gz //解压tar.gz
# count md5 value,,查看文件是否完整
cd 01raw_data
ls
md5sum *gz>md5.txt
cat md5.txt
ls
md5sum -c md5.txt

MD5值检测
2.质量控制

质量控制
ls *gz |xargs -I [] echo 'nohup fastqc [] &'>fastqc.sh
bash fastqc.sh

Fastqc 碱基质量分布图
横坐标代表每个每个碱基的位置,反映了读长信息,比如测序的读长为150bp,横坐标就是1到150;
纵坐标代表碱基质量值,
图中的箱线图代表在每个位置上所有碱基的质量值分布,
中间的红线代表的是中位数
用黄色填充的区域的上下两端分别代表上四分位数和下四分位数;
箱线图最上方的短线代表90%,最下方的短线代表10%
蓝色的线代表平均值
背景色从上到在下依次为green, orange, red; 分别代表very good, reasonable, poor;将碱基质量分成3个不同的标准
当有一个位置的10%四分位数小于10或者中位数小于25时会给出警告;
当有一个位置的10%四分位数小于5或者中位数小于20时会提示失败;
如下图所示:
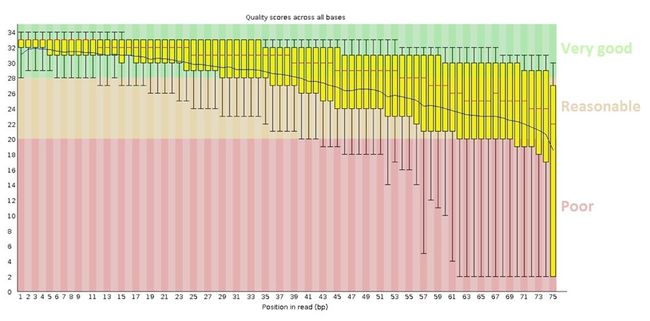
从上面图中我们可以看出读长为75bp;前15bp左右测序质量非常好;
随着测序的进行,由于试剂的消耗等原因,测序质量开始逐渐降低,最后的60-75bp质量就非常的差;
质量过滤


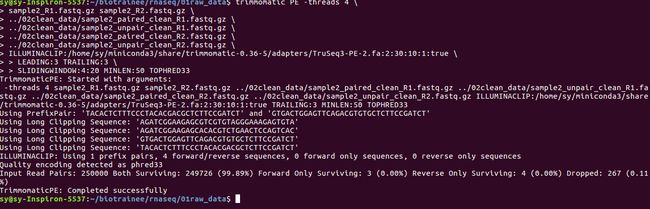
#以一个样品的两个重复 为例;
sample1_1_R1.fastq.gz sample1_1_R2.fastq.gz \
trimmomatic PE -threads 4 \
sample1_1_R1.fastq.gz sample1_1_R2.fastq.gz \
../02clean_data/sample1_1_paired_clean_R1.fastq.gz \
../02clean_data/sample1_1_unpair_clean_R1.fastq.gz \
../02clean_data/sample1_1_paired_clean_R2.fastq.gz \
../02clean_data/sample1_1_unpair_clean_R2.fastq.gz \
ILLUMINACLIP:/home/miniconda3/share/trimmomatic-0.36-5/adapters/TruSeq3-PE-2.fa:2:30:10:1:true \
LEADING:3 TRAILING:3 \
SLIDINGWINDOW:4:20 MINLEN:50 TOPHRED33
sample1_2_R1.fastq.gz sample1_2_R2.fastq.gz \
trimmomatic PE -threads 10 \
sample1_2_R1.fastq.gz sample1_2_R2.fastq.gz \
../02clean_data/sample1_2_paired_clean_R1.fastq.gz \
../02clean_data/sample1_2_unpair_clean_R1.fastq.gz \
../02clean_data/sample1_2_paired_clean_R2.fastq.gz \
../02clean_data/sample1_2_unpair_clean_R2.fastq.gz \
ILLUMINACLIP:/home/miniconda3/share/trimmomatic-0.36-5/adapters/TruSeq3-PE-2.fa:2:30:10:1:true \
LEADING:3 TRAILING:3 \
SLIDINGWINDOW:4:20 MINLEN:50 TOPHRED33
3.序列比对
STAR+RSEM 先比对再进行定量


# build index 建立索引;
STAR --runThreadN 10 --runMode genomeGenerate \
--genomeDir arab_STAR_genome \
--genomeFastaFiles 00ref/TAIR10_Chr.all.fasta \
--sjdbGTFfile 00ref/Araport11_GFF3_genes_transposons.201606.gtf \
--sjdbOverhang 149 # 一般为reads长度-1(150-1)

序列比对排序
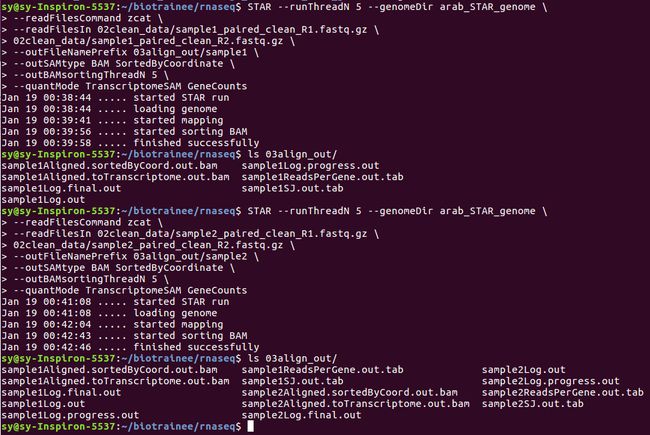
# STAR align 简单版本 样本R1
STAR --runThreadN 10 --genomeDir arab_STAR_genome \ #索引目录
--readFilesCommand zcat \ #执行解压缩:
--readFilesIn 02clean_data/sample1_1_paired_clean_R1.fastq.gz \
02clean_data/sample1_1_paired_clean_R2.fastq.gz \ #执行输入文件
--outFileNamePrefix 03align_out/sample1_1 \ #输出文件加前缀;
--outSAMtype BAM SortedByCoordinate \ #指定输出bam 文件并排序;
--outBAMsortingThreadN 5 \ #设置排序中的线程数;
--quantMode TranscriptomeSAM GeneCounts #将上面基因组比对的信息转化为转录本比对的信息,count
# --quantMode TranscriptomeSAM will output alignments translated into tran-script coordinates,为了使用RSEM 进行定量分析做准备;
# STAR align 复杂版本 样本R2
STAR --runThreadN 12 --genomeDir arab_STAR_genome \
--readFilesCommand zcat \
--readFilesIn 02clean_data/sample2_1_paired_clean_R1.fastq.gz \
02clean_data/sample2_1_paired_clean_R2.fastq.gz \
--outFileNamePrefix 03align_out/sample2_1 \
--outSAMtype BAM SortedByCoordinate \
--outBAMsortingThreadN 5 \
--quantMode TranscriptomeSAM GeneCounts
#less 查看文件比对信息
less **.final.out

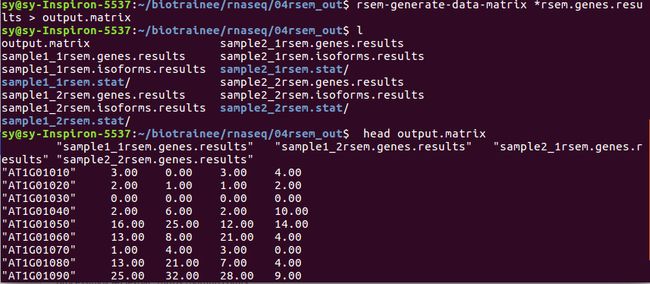
4.定量
测试rsem是否安装

2.png
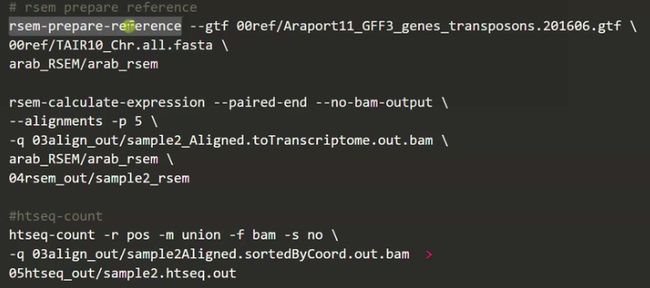
#samtools 查看bam 文件;(每个基因上有多少个reads 的统计信息的bam文件;)
#为什么tab文件中后面有三个文件;
从原始的read信息到比对文件的过程;
samtools view sample2_Aligned.sortedByCoord.out.bam |head
转录组数据计算表达定量;
# rsem prepare reference 从参考基因组中提取原始准备文件
rsem-prepare-reference --gtf 00ref/Araport11_GFF3_genes_transposons.201606.gtf \
00ref/TAIR10_Chr.all.fasta \
arab_RSEM/arab_rsem

RSEM建立索引
#sample
rsem-calculate-expression --paired-end --no-bam-output \
--alignments -p 15 \
-q 03align_out/sample1_1Aligned.toTranscriptome.out.bam \
arab_RSEM/arab_rsem \
04rsem_out/sample1_1rsem

RSEM进行定量
基因组,转录本表达定量结果

查看RSEM定量结果
基因表达定量方案选择
featureCount定量

featureCount
###featurecount
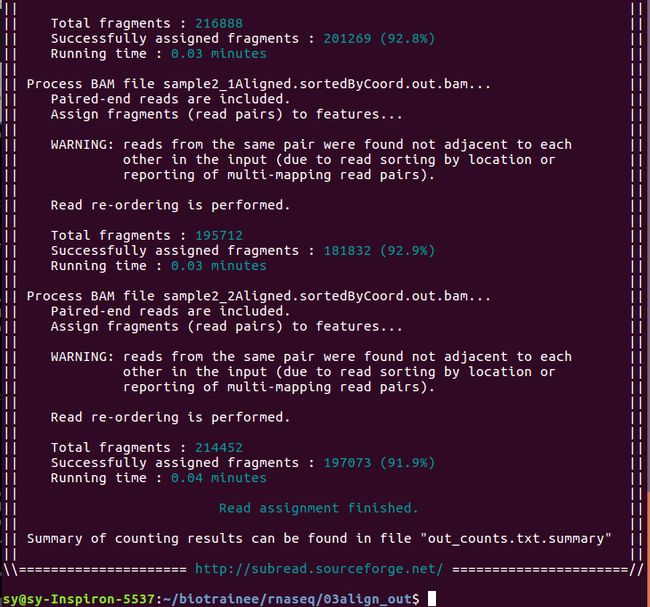
## programname -p(双端) -a (基因组注释文件) -o (输出文件) -T(使用的线程数) -t -g 通过什么来进行定量,输出什么结果 所有要定量的文件;
featureCounts -p -a ../00ref/Araport11_GFF3_genes_transposons.201606.gtf -o out_counts.txt -T 6 -t exon -g gene_id sample*Aligned.sortedByCoord.out.bam
featureCounts -p -a ../00ref/genes.gtf -o Z-13-1_out_counts.txt -T 10 -t exon -g *L1*dm6_Aligned.sortedByCoord.out.bam
featureCounts -p -a ../00ref/genes.gtf -o out_counts.txt -T 20 -t exon -g gene_id
featureCounts -p -a ../00ref/genes.gtf -o out_counts.txt -T 6 -t exon -g gene_id zgq-*_accepted_hits.bam

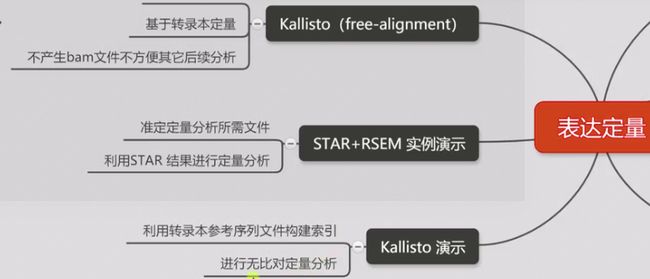
方案
kallisto 不比对,直接定量
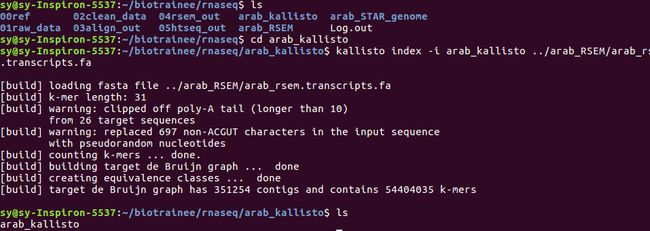
#kallisto 先构建索引 index; /arab_kallisto目录下
kallisto index -i arab_kallisto ../arab_RSEM/arab_rsem.transcripts.fa
#kallisto进行定量
kallisto quant -i Dro_kallisto/Dro_kallisto -o 05kallisto_out/sample2 02clean_data/R1.gz 02clean_data/R2.gz
kallisto quant -i Dro_kallisto/Dro_kallisto -o 05kallisto_out/Z-1 ../../Data_cy/20181018-embryo-RIP-seq\ data/Z-13-1_L1_310310.R1.clean.fastq.gz ../../Data_cy/20181018-embryo-RIP-seq\ data/Z-13-1_L1_310310.R2.clean.fastq.gz
#abundance.tsv 列:gene counts TPM
kallisto建立索引
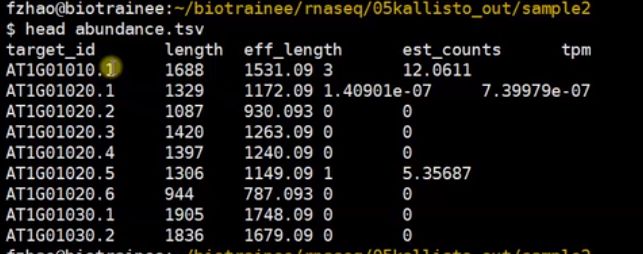
kallisto定量结果
5.差异基因分析:
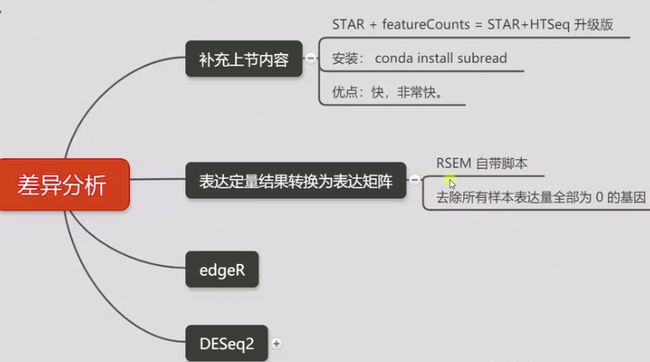
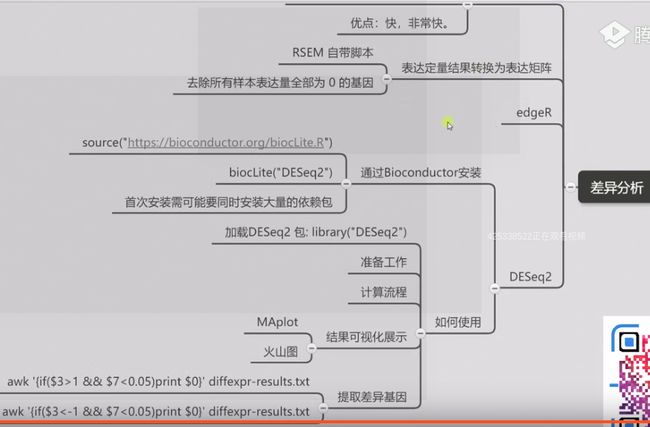
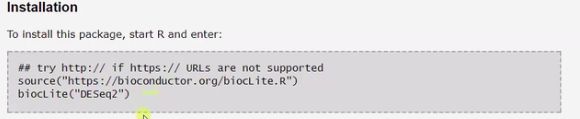
R install
install.packages("ggrepel")
install.packages("ggplot2")
### 差异表达
mkdir 06deseq_out
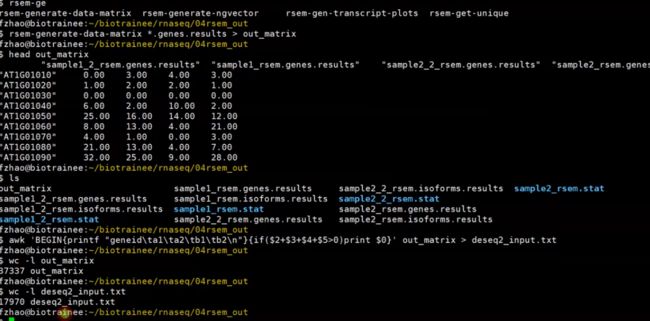
#构建表达矩阵
rsem-generate-data-matrix *rsem.genes.results > output.matrix
矩阵合并
#删除未检测到表达的基因
awk 'BEGIN{printf "geneid\ta1\ta2\tb1\tb2\n"}{if($2+$3+$4+$5>0)print $0}' output.matrix > deseq2_input.txt
###查看文件的行数
wc -l output.matrix
wc -l deseq2_input.txt
mv deseq2_input.txt ../06deseq_out/
mv deseq2.r ../06deseq_out/
abundance_estimates_to_matrix.pl
run_DE_analysis.pl
差异基因结果
差异基因作图

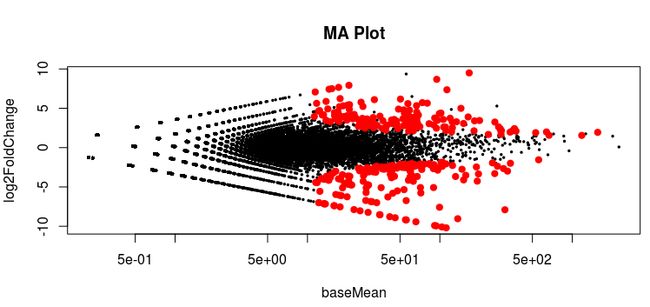
MAplot

火山图
R脚本
### 设置当前目录;
打开一个文件夹中的R脚本,session
或 输入路径;setwd("~/00")
# read.table ,文件有header,第一行为row.name;
input_data <- read.table("deseq2_input.txt", header=TRUE, row.names=1)
# 取整,函数round
input_data <-round(input_data,digits = 0)
# 准备文件
# as.matrix 将输入文件转换为表达矩阵;
input_data <- as.matrix(input_data)
# 控制条件:因子(对照2,实验2)
condition <- factor(c(rep("ct1", 2), rep("exp", 2)))
# input_data根据控制条件构建data.frame
coldata <- data.frame(row.names=colnames(input_data), condition)
library(DESeq2)
# build deseq input matrix 构建输入矩阵
#countData作为矩阵的input_data;colData Data.frame格式;控制条件design;
dds <- DESeqDataSetFromMatrix(countData=input_data,colData=coldata, design=~condition)
# DESeq2 进行差异分析
dds <- DESeq(dds)
#实际包含3步,请自行学习;
# 提取结果
#dds dataset格式转换为DESeq2 中result数据格式,矫正值默认0.1
res <- results(dds,alpha=0.05)
#查看res(DESeqresults格式),可以看到上下调基因
summary(res)

上下调基因
#res(resultset)按照校正后的P值排序
res <- res[order(res$padj), ]
res
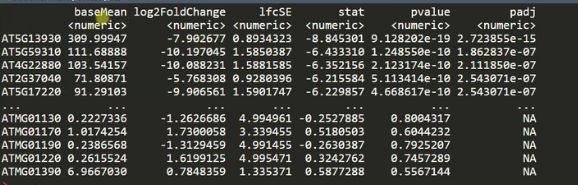
# 将进过矫正后的表达量结果加进去;
resdata <- merge(as.data.frame(res), as.data.frame(counts(dds, normalized=TRUE)),by="row.names",sort=FALSE)
names(resdata)[1] <- "Gene"
#查看(resdata)
# output result 输出结果
write.table(resdata,file="diffexpr-results.txt",sep = "\t",quote = F, row.names = F)
#可视化展示
plotMA(res)
maplot <- function (res, thresh=0.05, labelsig=TRUE,...){
with(res,plot(baseMean, log2FoldChange, pch=20, cex=.5, log="x", ...))
with(subset(res, padj 1)
ggplot(data=resdata, aes(x=log2FoldChange, y =-log10(padj),color =significant)) +
geom_point() +
ylim(0, 8)+
scale_color_manual(values =c("black","red"))+
geom_hline(yintercept = log10(0.05),lty=4,lwd=0.6,alpha=0.8)+
geom_vline(xintercept = c(1,-1),lty=4,lwd=0.6,alpha=0.8)+
theme_bw()+
theme(panel.border = element_blank(),
panel.grid.major = element_blank(),
panel.grid.minor = element_blank(),
axis.line = element_line(colour = "black"))+
labs(title="Volcanoplot_biotrainee (by sunyi)", x="log2 (fold change)",y="-log10 (padj)")+
theme(plot.title = element_text(hjust = 0.5))+
geom_text_repel(data=subset(resdata, -log10(padj) > 6), aes(label=Gene),col="black",alpha =0.8)
差异基因的提取
#查看符合条件的差异基因
awk '{if($3>1 && $7<0.05)print $0}' diffexpr-results.txt |head
#查看差异基因有多少行
awk '{if($3>1 && $7<0.05)print $0}' diffexpr-results.txt |wc -l
#提取某几列
awk '{if($3>1 && $7<0.05)print $0}' diffexpr-results.txt |cut -f 1,2,4,7 |head
#上调基因的提取;
awk '{if($3>1 && $7<0.05)print $0}' diffexpr-results.txt |cut -f 1,2,4,7 > up.gene.txt
#下调基因的提取;
awk '{if($3<-1 && $7<0.05)print $0}' diffexpr-results.txt |cut -f 1,2,4,7 > down.gene.txt
根据第1列是Geneid,第7,8,9,10列是counts数,用awk提取出geneID和counts。
awk -F '\t' '{print $1,$7,$8,$9,$10}' OFS='\t' out_counts.txt >subread_matrix.out