刘小泽写于19.8.19
主要介绍官方教程PBMC 3K的下游分析(https://satijalab.org/seurat/v3.0/pbmc3k_tutorial.html)
上篇:单细胞Seurat包升级之2,700 PBMCs分析(上)
数据标准化(scale)
它的目的是实现数据的线性转换(scaling),这也是降维处理(如PCA)之前的标准预处理。
主要利用了ScaleData函数,回忆一下,之前归一化(normalize)这里做的操作是log处理,它是对所有基因的表达量统一对待的,最后放在了一个起跑线上。但是为了真正找到差异,我们还要基于这个起跑线,考虑不同样本对表达量的影响
它做了:
- 将每个基因在所有细胞的表达量进行调整,使得每个基因在所有细胞的表达量均值为0
- 将每个基因的表达量进行标准化,让每个基因在所有细胞中的表达量方差为1
# 先使用全部基因
all.genes <- rownames(pbmc)
> length(all.genes)
[1] 13714
pbmc <- ScaleData(pbmc, features = all.genes)
# 结果存储在pbmc[["RNA"]]@scale.data中
感觉这一步太慢,能不能加速一点?
这一步主要目的就是下一步的降维,使用全部基因是比较慢。如果想提高速度,可以只对鉴定的HVGs(之前FindVariableFeatures设置的是2000个)进行操作,其实这个函数默认就是对部分差异基因进行操作,:pbmc <- ScaleData(pbmc) 。这样操作的结果中降维和聚类不会被影响,但是只对HVGs基因进行标准化,下面画的热图依然会受到全部基因中某些极值的影响。所以为了热图的结果,还是对所有基因进行标准化比较好。
ScaleData的操作过程是怎样的?
看一下帮助文档就能了解:
ScaleData这个函数有两个默认参数:do.scale = TRUE和do.center = TRUE,然后需要输入进行scale/center的基因名(默认是取前面FindVariableFeatures的结果)。
scale和center这两个默认参数应该不陌生了,center就是对每个基因在不同细胞的表达量都减去均值;
scale就是对每个进行center后的值再除以标准差(就是进行了一个z-score的操作)
再看一个来自BioStar的讨论:https://www.biostars.org/p/349824/
问题1:为什么在NormalizeData()之后,还需要进行ScaleData操作?
前面NormalizeData进行的归一化主要考虑了测序深度/文库大小的影响(reads *10000 divide by total reads, and then log),但实际上归一化的结果还是存在基因表达量分布区间不同的问题;而ScaleData做的就是在归一化的基础上再添zero-centres (mean/sd =》 z-score),将各个表达量放在了同一个范围中,真正实现了”表达量的同一起跑线“。
另外,新版的ScaleData函数还支持设定回归参数,如(nUMI、percent.mito),来校正一些技术噪音
问题2: FindVariableGenes() or RunPCA() or FindCluster()这些参数是基于归一化Normalized_Data还是标准化Scaled_Data ?
所有操作都是基于标准化的数据Scaled_Data ,因为这个数据是针对基因间比较的。例如有两组基因表达量如下:
g1 10 20 30 40 50
g2 20 40 60 80 100
虽然它们看起来是g2的表达量是g1的两倍,但真要降维聚类时,就要看scale的结果:
> scale(c(10,20,30,40,50))
[,1]
[1,] -1.2649111
[2,] -0.6324555
[3,] 0.0000000
[4,] 0.6324555
[5,] 1.2649111
attr(,"scaled:center")
[1] 30
attr(,"scaled:scale")
[1] 15.81139
> scale(c(20,40,60,80,100))
[,1]
[1,] -1.2649111
[2,] -0.6324555
[3,] 0.0000000
[4,] 0.6324555
[5,] 1.2649111
attr(,"scaled:center")
[1] 60
attr(,"scaled:scale")
[1] 31.62278
二者标准化后的分布形态是一样的(只是中位数不同),而我们后来聚类也是看数据的分布,分布相似聚在一起。所以归一化差两倍的两个基因,根据标准化的结果依然聚在了一起
问题3: ScaleData()在scRNA分析中一定要用吗?
实际上标准化这个过程不是单细胞分析固有的,在很多机器学习、降维算法中比较不同feature的距离时经常使用。当不进行标准化时,有时feature之间巨大的差异(就像问题2中差两倍的基因表达量,实际上可能差的倍数会更多)会影响分析结果,让我们误以为它们的数据分布相差很远。
很多时候,我们会混淆normalization和scale:那么看一句英文解释:
Normalization "normalizes" within the cell for the difference in sequenicng depth / mRNA throughput. 主要着眼于样本的文库大小差异
Scaling "normalizes" across the sample for differences in range of variation of expression of genes . 主要着眼于基因的表达分布差异
另外,看scale()函数的帮助文档就会发现,它实际上是对列进行操作:

那么具体操作中是要先对表达矩阵转置,然后scale,最后转回来
t(scale(t(dat[genes,])))
问题4:RunCCA()函数需要归一化数据还是标准化数据?
通过看这个函数的帮助文档:
RunCCA(object, object2, group1, group2, group.by, num.cc = 20, genes.use,
scale.data = TRUE, rescale.groups = FALSE, ...)
显示scale.data = TRUE,那么就需要标准化后的数据
怎样移除不想要的差异来源?
在进行降维聚类时,主要就是考虑数据差异对分布产生的影响。由于技术误差导致的差异是我们不感兴趣的,排除这一部分其实也在上面的问题1中提到了,如果说不想线粒体污染导致的差异影响整体差异分布:
pbmc <- ScaleData(pbmc, vars.to.regress = "percent.mt")
这个函数仅针对初级用户,如果想深入探索技术误差的排除,官方给出了另一个专业版函数:SCTransform,它也包含vars.to.regress这个选项。详细的说明在:https://satijalab.org/seurat/v3.0/sctransform_vignette.html
进行线性降维
最常用的线性降维就是PCA方法,使用标准化的数据
它默认选择之前鉴定的差异基因(2000个)作为input,但可以使用features进行设置;默认分析的主成分数量是50个
# 这里就使用默认值
pbmc <- RunPCA(pbmc, features = VariableFeatures(object = pbmc))
# 结果保存在reductions 这个接口中
检查下PCA结果:
> print(pbmc[["pca"]], dims = 1:3, nfeatures = 5)
# 或者用 print(pbmc@reductions$pca, dims = 1:3, nfeatures = 5)
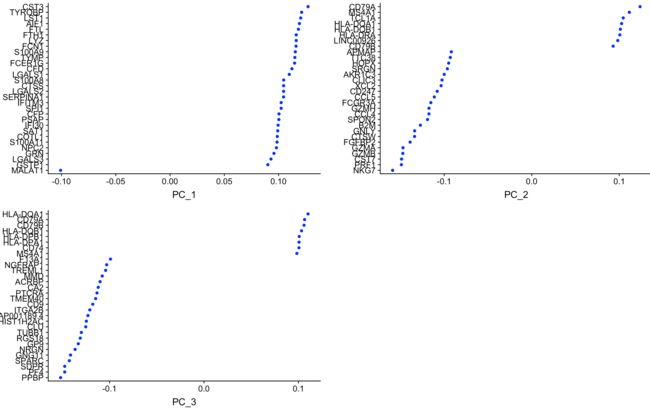
PC_ 1
Positive: CST3, TYROBP, LST1, AIF1, FTL
Negative: MALAT1, LTB, IL32, IL7R, CD2
PC_ 2
Positive: CD79A, MS4A1, TCL1A, HLA-DQA1, HLA-DQB1
Negative: NKG7, PRF1, CST7, GZMB, GZMA
PC_ 3
Positive: HLA-DQA1, CD79A, CD79B, HLA-DQB1, HLA-DPB1
Negative: PPBP, PF4, SDPR, SPARC, GNG11
用VizDimLoadings对print的结果可视化:
VizDimLoadings(pbmc, dims = 1:3, reduction = "pca")
用DimPlot进行降维后的可视化
提取两个主成分(默认前两个,当然可以修改dims选项)绘制散点图
DimPlot(pbmc, reduction = "pca")

这个图中每个点就是一个样本,它根据PCA的结果坐标进行画图,这个坐标保存在了cell.embeddings中:
> head(pbmc[['pca']]@cell.embeddings)[1:2,1:2]
PC_1 PC_2
AAACATACAACCAC -4.7296855 -0.5184265
AAACATTGAGCTAC -0.5174029 4.5918957
其实还支持定义分组:根据[email protected]中的ident分组:
> table([email protected]$orig.ident)
pbmc3k
2638
# 当然这里只有一个分组
探索异质性来源—DimHeatmap
每个细胞和基因都根据PCA结果得分进行了排序,默认画前30个基因(nfeatures设置),1个主成分(dims设置),细胞数没有默认值(cells设置)
DimHeatmap(pbmc, dims = 1:15, cells = 500, balanced = TRUE)
# 其中balanced表示正负得分的基因各占一半
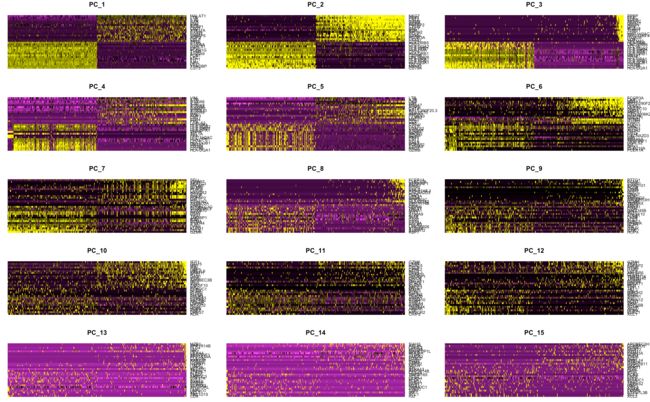
降维的真实目的是尽可能减少具有相关性的变量数目,例如原来有700个样本,也就是700个维度/变量,但实际上根据样本中的基因表达量来归类,它们或多或少都会有一些关联,这样有关联的一些样本就会聚成一小撮,表示它们的”特征距离“相近。最后直接分析这些”小撮“,就用最少的变量代表了实际最多的特征
既然每个主成分都表示具有相关性的一小撮变量(称之为”metafeature“,元特征集),那么问题来了:
降维后怎么选择合适数量的主成分来代表整个数据集?
选10个?20个?50个?最简单的方法就是:
ElbowPlot(pbmc)
它是根据每个主成分对总体变异水平的贡献百分比排序得到的图,我们主要关注”肘部“的PC,它是一个转折点(也即是这里的PC9-10),说明取前10个主成分可以比较好地代表总体变化
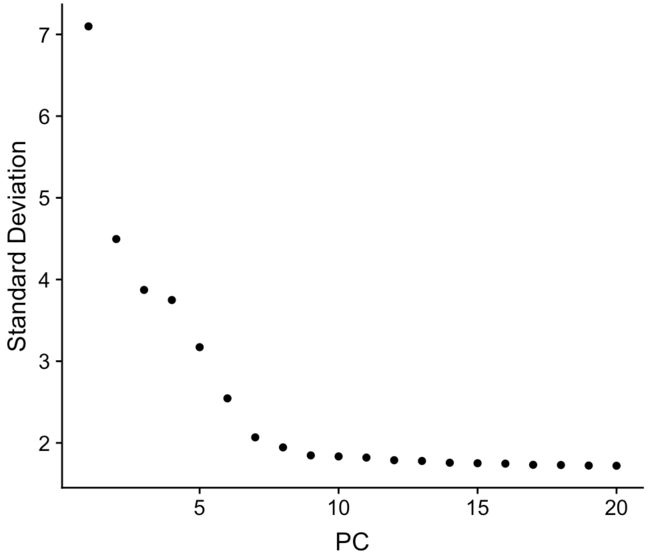
但官方依然建议我们,下游分析时多用几个成分试试(10个、15个甚至50个),如果起初选择的合适,结果不会有太大变化;另外推荐开始不确定时要多选一些主成分,也不要直接就定下5个成分进行后续分析,那样很有可能会出错
细胞聚类
Seurat 3版本提供了graph-based 聚类算法,参考两篇文章:SNN-Cliq, Xu and Su, Bioinformatics, 2015] 和 PhenoGraph, Levine et al., Cell, 2015]. 简言之,这些graph的方法都是将细胞嵌入一个算法图层中,例如:K-nearest neighbor (KNN) graph 中,每个细胞之间的连线就表示相似的基因表达模式,然后按照这个相似性将图分隔成一个个的高度相关的小群体(名叫‘quasi-cliques’ or ‘communities’);

又比如,在PhenoGraph中,首先根据PCA的欧氏距离结果,构建了KNN图,找到了各个近邻组成的小社区;然后根据近邻中两两住户(细胞)之间的交往次数(shared overlap)计算每条线的权重(术语叫Jaccard similarity) 。这些计算都是用包装好的函数FindNeighbors() 得到的,它的输入就是前面降维最终确定的主成分
# 因为我们前面挑选了10个PCs,所以这里dims定义为10个
pbmc <- FindNeighbors(pbmc, dims = 1:10)
以上是凭个人理解发挥,大概就是这么个意思,不涉及算法层面推导
得到权重后,为了对细胞进行聚类,使用了计算模块的算法(默认使用Louvain,另外还有包括SLM的其他三种),使用FindClusters() 进行聚类。其中包含了一个resolution的选项,它会设置一个”间隔“值,该值越大,间隔越大,得到的cluster数量越多
怎么理解这个
resolution参数?
可以想象成配眼镜的时候,镜片度数越高,分辨率越高,看的越清楚,看到的细节越丰富(cluster越多);反之,如果分辨率调的很低,结果就看的模模糊糊一大坨
一般来说,这个值在细胞数量为3000左右时设为0.4-1.2 会有比较好的结果
pbmc <- FindClusters(pbmc, resolution = 0.5)
# 结果聚成几类,用Idents查看
> head(Idents(pbmc), 5)
AAACATACAACCAC AAACATTGAGCTAC AAACATTGATCAGC AAACCGTGCTTCCG
1 3 1 2
AAACCGTGTATGCG
6
Levels: 0 1 2 3 4 5 6 7 8
# 发现3000细胞,设resolution=0.5时,得到Number of communities: 9
另一种降维方法—非线性降维(UMAP/tSNE)
线性降维PCA(1933)一个特点就是:它将高维中不相似的数据点放在较低维度时,会尽可能多地保留原始数据的差异,因此导致这些不相似的数据相距甚远,大多的数据重叠放置
tSNE(2008)兼顾了高维降维+低维展示,降维后的那些不相似的数据点依然可以放得靠近一些,保证了点既在局部分散,又在全局聚集
后来开发的UMAP(2018)相比tSNE又能展示更多的维度(参考文献:https://sci-hub.tw/https://doi.org/10.1038/nbt.4314)
做个对比:

# 使用UMAP
pbmc <- RunUMAP(pbmc, dims = 1:10)
DimPlot(pbmc, reduction = "umap") # 还可以设置label = TRUE让数字显示在每个cluster上
# 对计算过程很费时的结果保存一下
save(pbmc, file = 'pbmc_umap.Rdata')
需要注意的点:
注意1:tSNE算法具有随机性,为了保证重复性,要固定一个随机种子
注意2:如何在R中使用UMAP?
以下测试是在个人电脑上,需要管理员权限
因为UMAP是基于Python的,如果要用它,必须先保证有一个python的安装环境,先试验一下:
> reticulate::py_install(packages ='umap-learn')
# 结果报错,说让我升级一下virtualenv(mac和linux默认使用virtualenv,而Windows只能使用conda)
Error: Prerequisites for installing Python packages not available.
Execute the following at a terminal to install the prerequisites:
$ sudo /usr/local/bin/pip install --upgrade virtualenv
然后直接在Rstudio中打开Terminal,输入:sudo /usr/local/bin/pip install --upgrade virtualenv
再运行一遍上面的R代码(成功与否取决于个人的网络,因为它要通过外链去下载,wlan有限制的话就用个人热点吧):
一般总共二三十兆的文件,需要下载个十几分钟,速度确实很慢,而这个过程很有可能出现断线,只能重新运行安装。已下载完的文件会直接识别不会重复下载。检查是否下载成功标志就是:
library(umap)能否成功,这个是Seurat调用的基础。最后特别注意,要重启一下Rstudio再运行RunUMAP
找差异表达基因(cluster biomarker)
目的是根据表达量不同这个特征而分出不同细胞群的基因们(就是找具有生物学意义的HVGs=》即biomarkers)
marker可以理解成标记,就是一看到有这些基因,就说明是这个细胞群体
主要利用FindAllMarkers()函数,它默认会对单独的一个细胞群与其他几群细胞进行比较找到正、负表达biomarker(这里的正负有点上调、下调基因的意思;正marker表示在我这个cluster中表达量高,而在其他的cluster中低)
# 找到cluster1中的marker基因
cluster1.markers <- FindMarkers(pbmc, ident.1 = 1, min.pct = 0.25)
head(cluster1.markers, n = 5)
上面这个代码中min.pct的意思是:一个基因在任何两群细胞中的占比最低不能低于多少,当然这个可以设为0,但会大大加大计算量
另外,如果要比较cluster5和cluster0、cluster3的marker基因:
cluster5.markers <- FindMarkers(pbmc, ident.1 = 5, ident.2 = c(0, 3), min.pct = 0.25)
head(cluster5.markers, n = 5)
一步到位的办法FindAllMarkers(对所有cluster都比较一下,并只挑出来正表达marker):
pbmc.markers <- FindAllMarkers(pbmc, only.pos = TRUE, min.pct = 0.25, logfc.threshold = 0.25)
# 这一步过滤好好理解(进行了分类、排序、挑前2个)
pbmc.markers %>% group_by(cluster) %>% top_n(n = 2, wt = avg_logFC)
Seurat提供了多种差异分析的检验方法,在test.use选项中设置(详见:DE vignette )例如:
cluster1.markers <- FindMarkers(pbmc, ident.1 = 0, logfc.threshold = 0.25, test.use = "roc", only.pos = TRUE)
多种可视化方法:
-
VlnPlot:用小提琴图对某些基因在全部cluster中表达量进行绘制VlnPlot(pbmc, features = c("MS4A1", "CD79A"))
-
FeaturePlot:最常用的可视化=》将基因表达量投射到降维聚类结果中FeaturePlot(pbmc, features = c("MS4A1", "GNLY", "CD3E", "CD14", "FCER1A", "FCGR3A", "LYZ", "PPBP", "CD8A"))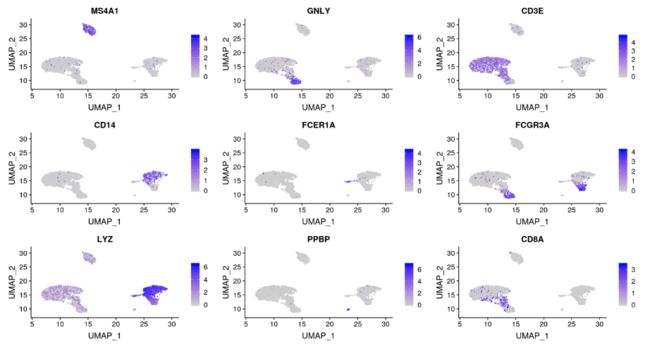
另外还有:RidgePlot, CellScatter, and DotPlot
-
DoHeatmap对给定细胞和基因绘制热图,例如对每个cluster绘制前20个markertop10 <- pbmc.markers %>% group_by(cluster) %>% top_n(n = 10, wt = avg_logFC) DoHeatmap(pbmc, features = top10$gene) + NoLegend()
赋予每个cluster细胞类型
重点在于背景知识的理解,能够将marker与细胞类型对应起来,例如这里从各个cluster中找到的marker对应到了不同的细胞(这一过程是比较考验课题理解程度的):
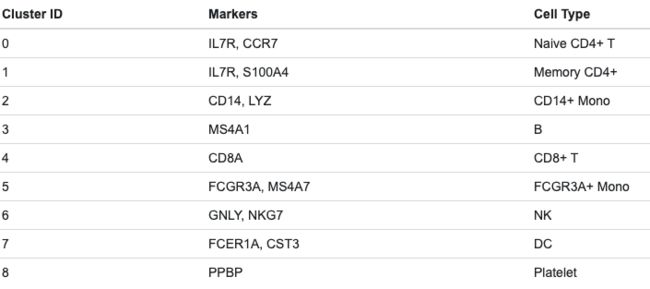
有了这个,绘图就很简单:
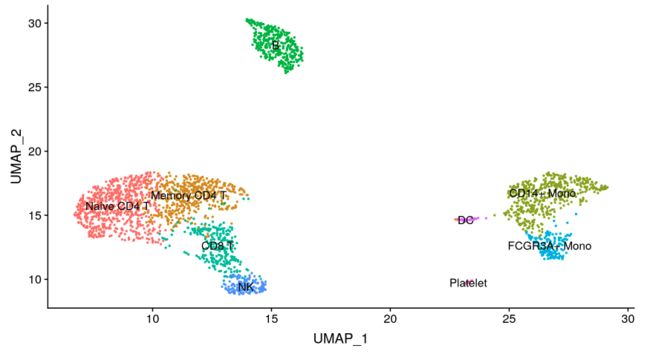
new.cluster.ids <- c("Naive CD4 T", "Memory CD4 T", "CD14+ Mono", "B", "CD8 T", "FCGR3A+ Mono", "NK", "DC", "Platelet")
names(new.cluster.ids) <- levels(pbmc)
pbmc <- RenameIdents(pbmc, new.cluster.ids)
DimPlot(pbmc, reduction = "umap", label = TRUE, pt.size = 0.5) + NoLegend()
欢迎关注我们的公众号~_~
我们是两个农转生信的小硕,打造生信星球,想让它成为一个不拽术语、通俗易懂的生信知识平台。需要帮助或提出意见请后台留言或发送邮件到[email protected]
