在bioconductor上面看到一个R包 seqCNA:
- R Script
读其文档的时候发现,是可以针对单个样本进行拷贝数变异分析的,代码如下:
library(seqCNA)
data(seqsumm_HCC1143)
head(seqsumm_HCC1143)
dim(seqsumm_HCC1143)
tail(seqsumm_HCC1143)
## 200Kb windows to calculate the GC content and counts.
rco = readSeqsumm(tumour.data=seqsumm_HCC1143)
rco = applyFilters(rco, trim.filter=1, mapq.filter=2)
rco = runSeqnorm(rco)
rco = runGLAD(rco)
plotCNProfile(rco)
rco = applyThresholds(rco, seq(-0.8,4,by=0.8), 1)
plotCNProfile(rco)
summary(rco)
head(rco@output)
writeCNProfile(rco,'./')
虽然其算法比较复杂,但是用法很简单,对input的数据进行了多步骤处理,而且其input数据本身是比较简单的,如下:
> head(seqsumm_HCC1143)
chrom win.start reads.gc reads.mapq counts
1 chr1 0e+00 0.551 1.691 1199
2 chr1 2e+05 0.534 0.620 824
3 chr1 4e+05 0.457 0.831 8469
4 chr1 6e+05 0.545 6.479 1459
5 chr1 8e+05 0.720 31.619 1094
6 chr1 1e+06 0.766 38.205 976
> dim(seqsumm_HCC1143)
[1] 5314 5
> tail(seqsumm_HCC1143)
chrom win.start reads.gc reads.mapq counts
5309 chr5 179800000 0.559 35.081 1946
5310 chr5 180000000 0.568 35.668 1970
5311 chr5 180200000 0.545 34.427 1790
5312 chr5 180400000 0.572 34.286 1569
5313 chr5 180600000 0.586 22.844 1591
5314 chr5 180800000 0.562 0.319 845
就是对单个样本的bam文件进行200kb的窗口进行滑动计算每个窗口的gc含量,该窗口区域覆盖的reads数量,还有比对的质量值,很容易写脚本进行计算。
GENOME='/home/jianmingzeng/reference/genome/human_g1k_v37/human_g1k_v37.fasta'
bam='ESCC13-T1_recal.bam'
samtools mpileup -f $GENOME $bam |\
perl -alne '{$pos=int($F[1]/200000); $key="$F[0]\t$pos";$GC{$key}++ if $F[2]=~/[GC]/;$counts_sum{$key}+=$F[3];$number{$key}++;}END{print "$_\t$number{$_}\t$GC{$_}\t$counts_sum{$_}" foreach keys %number}' |\
sort -k1,1 -k 2,2n >T1.windows
得到的结果如下:
head GC_stat.10k.txt
chr1 1 7936 3885 582219
chr1 2 2123 934 88167
chr1 3 1969 169 28729
chr1 4 3556 593 48724
chr1 5 8828 2582 176627
chr1 6 8229 2290 117675
chr1 7 8794 438 156158
chr1 8 10000 723 211816
chr1 9 9077 2421 247285
chr1 10 9415 1661 371830
前面两行是窗口的坐标,第几号染色体的第几个窗口,后面3行是数据,分别是每个窗口的测到的碱基数,GC碱基数,测序总深度。
然后走seqCNA流程
library(seqCNA)
a=read.table('wgs/GC_stat.10k.txt',fill = T)
a=na.omit(a)
a$GC = a[,4]/a[,3]
a$depth = a[,5]/a[,3]
a = a[a$depth<100,]
plot(a$GC,a$depth)
a=a[,c(1,2,6,5)]
colnames(a)=c('chrom','win.start','reads.gc','counts')
a$counts=floor(a$counts/150)
a$reads.mapq=30
library(seqCNA)
head(a)
dim(a)
tail(a)
a$win.start=a$win.start*10000
a=a[a$chrom %in% paste0('chr',c(1:22,'X','Y')),]
a=a[a$win.start>0,]
a=a[a$counts>0,]
a=a[a$reads.gc>0,]
a=a[,c(1:3,5,4)]
## 200Kb windows to calculate the GC content and counts.
rco = readSeqsumm(tumour.data=a)
rco = applyFilters(rco, trim.filter=1, mapq.filter=2)
rco = runSeqnorm(rco)
rco = runGLAD(rco)
plotCNProfile(rco)
rco = applyThresholds(rco, seq(-0.8,4,by=0.8), 1)
plotCNProfile(rco)
summary(rco)
head(rco@output)
writeCNProfile(rco,'./')
分析结果汇总信息:
Basic information:
SeqCNAInfo object with 306397 10Kbp-long windows.
PEM information is not available.
Paired normal is not available.
Genome and build unknown (chromosomes chr1 to chrY).
Total filtered windows: 23717.
The profile is normalized and segmented.
Copy numbers called from 1 to 8.
CN1:34924 windows.
CN2:95226 windows.
CN3:116829 windows.
CN4:35047 windows.
CN5:554 windows.
CN6:100 windows.
CN7:0 windows.
CN8:0 windows.
单个样本的CNV结果如图
[图片上传失败...(image-c5e27f-1516675104040)]
[图片上传失败...(image-e2e6b8-1516675104040)]
[图片上传失败...(image-6fcea7-1516675104040)]
为什么要计算GC含量呢
这个是二代测序本身的技术限制,很容易探究到测序深度和GC含量是显著相关的,代码如下:
a=read.table('T1.windows')
a$GC = a[,4]/a[,3]
a$depth = a[,5]/a[,3]
a = a[a$depth<100,]
plot(a$GC,a$depth)
library(ggplot2)
# GET EQUATION AND R-SQUARED AS STRING
# SOURCE: http://goo.gl/K4yh
lm_eqn <- function(x,y){
m <- lm(y ~ x);
eq <- substitute(italic(y) == a + b %.% italic(x)*","~~italic(r)^2~"="~r2,
list(a = format(coef(m)[1], digits = 2),
b = format(coef(m)[2], digits = 2),
r2 = format(summary(m)$r.squared, digits = 3)))
as.character(as.expression(eq));
}
p=ggplot(a,aes(GC,depth)) + geom_point() +
geom_smooth(method='lm',formula=y~x)+
geom_text(x = 0.5, y = 100, label = lm_eqn(a$GC , a$depth), parse = TRUE)
p=p+theme_set(theme_set(theme_bw(base_size=20)))
p=p+theme(text=element_text(face='bold'),
axis.text.x=element_text(angle=30,hjust=1,size =15),
plot.title = element_text(hjust = 0.5) ,
panel.grid = element_blank(),
#panel.border = element_blank()
)
print(p)
可以很明显看到GC含量和测序深度是高度相关的
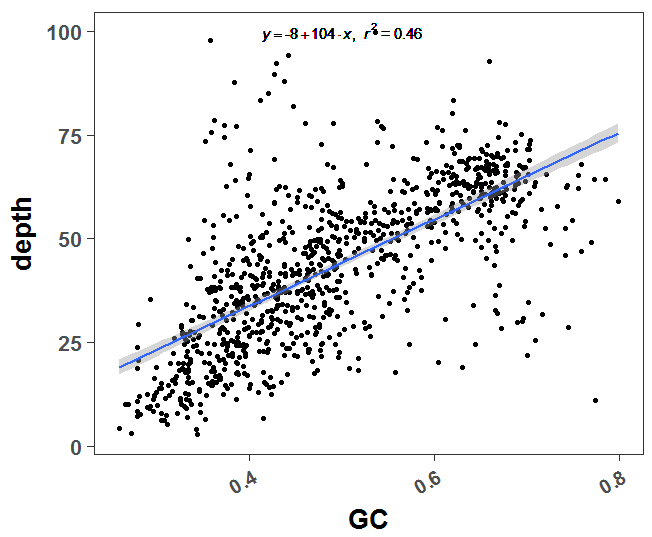
GC含量和测序深度