Gromacs——蛋白质-配体复合物教程翻译
1.准备
从RCSB网站下载T4溶菌酶pdb文件(代号3HTB),删除水。或下载已经准备好的pdb文件。
所用的配体为JZ4,即2-丙基苯酚,但它不能被Gromacs提供的力场识别,所以将分成两步准备拓扑文件:
a.用pdb2gmx准备蛋白质拓扑;
b.用其他工具准备配体拓扑。
所以需将蛋白质和JZ4保存到不同的坐标文件中。保存JZ4坐标:
grep JZ4 3HTB_clean.pdb>jz4.pdb
然后从3HTB_clean.pdb中删除JZ4行。
所用的力场是CHARMM36,可从MacKerell lab website下载。下载最新的CHARMM36力场压缩包和 "cgenff_charmm2gmx.py" 转换脚本(注意要与Python版本对应)。在工作目录中解压缩:
tar -zxvf charmm36-mar2019.ff.tgz
在工作目录中会出现 "charmm36-mar2019.ff"子目录。用pdb2gmx生成T4溶菌酶的拓扑文件:
gmx pdb2gmx -f 3HTB_clean.pdb -o 3HTB_processed.gro
选择默认的水模型和“当前目录中的”CHARRM36力场(1选项)。
对于OPLS、AMBER和CHARMM力场,力场不能识别的分子的参数推导通常需要量子力学计算。而对于Gromacs力场,则依赖于凝聚态行为的经验拟合。下表为一些计算多种力场参数的自动化程序,它们并不同样精确:

向JZ4添加H
本教程将用CGenFF服务器生成JZ4拓扑。访问上表中的链接,注册并激活。CGenFF需要Sybyl .mol2文件作为输入采集原子类型和键信息。CHARRM为全原子力场,所以必须有H.而晶体结构通常不包含H坐标,必须重建。用Avogadro程序产生.mol2文件并添加H原子。在Avogadro中打开jz4.pdb,从Build菜单中选择添加H.另存为jz4.mol2(选择Sybyl Mol2).
在用jz4.mol2之前必须进行修正:
第一个更改是在MOLECULE标题中。将“ *****”替换为“ JZ4”;
接下来,固定残基名称和编号,使它们都相同。该.mol2文件仅包含一个分子,因此应仅指定一个残基名称和编号。编辑后,jz4.mol2的更正ATOM部分应显示为:

最后,请注意@
perl sort_mol2_bonds.pl jz4.mol2 jz4_fix.mol2
下面的步骤用jz4_fix.mol2.
用CGenFF生产JZ4拓扑
登录CGenFF服务器的账号,点击“Upload molecule”,上传jz4_fix.mol2,会返回一个CHARMM流形式的拓扑文件(扩展名为.str).保存为jz4.str或从此处下载。
CHARMM流文件包含所有拓扑信息-原子类型,电荷和键连接。它还有一些附加的绑定参数部分,这些参数是通过类比为力场未涵盖的任何内部相互作用生成的。CGenFF还为每个参数提供惩罚分数,即评估分配的参数的可靠性。低于10的任何东西都被认为可以立即使用。介于10到50之间的值表示必须对拓扑进行一些验证,并且任何大于50的惩罚通常需要手动重新参数化。罚分是CGenFF服务器最重要的功能之一。许多其他服务器生成拓扑,它们是“黑匣子”,用户只需要信任即可。在没有验证的情况下将整个研究项目放在自动程序上非常危险。较差的配体拓扑会导致大量的时间浪费和不可靠的结果。始终验证新参数化物种的拓扑!至少要检查力场中现有部分的电荷大小和分配给配体的原子类型。
检查jz4.str的内容,并查看电荷和新的二面体参数的惩罚。它们都非常低,表明该拓扑的质量非常好,可以直接用于我们的仿真。
CHARMM格式的JZ4拓扑很好,但是如果我们尝试在GROMACS中运行仿真,它就没有用。使用我们先前从MacKerell网站下载的cgenff_charmm2gmx.py脚本。脚本名称将包含版本_py2或_py3,但为简单起见,此处省略了此信息,但您将需要使用为Python版本下载的脚本的实际名称。使用以下命令执行转换:
python cgenff_charmm2gmx.py JZ4 jz4_fix.mol2 jz4.str charmm36-mar2019.ff
请注意:此脚本需要NetworkX软件包,版本1.11-与NetworkX 2.x不兼容,并且将退出并显示明显错误。
成功哦转换后会出现下列输出:
============ DONE ============
Conversion complete.
The molecule topology has been written to jz4.itp
Additional parameters needed by the molecule are written to jz4.prm, which needs to be included in the system .top
============ DONE ============该配体引入了新的结合参数,这些参数不是现有力场的一部分,并且将这些参数写入到名为“ jz4.prm”的文件中,该文件采用GROMACS .itp文件的格式。我们将在短期内处理此文件,但重要的是要注意它的存在。现在将配体拓扑写入“ jz4.itp”,其中包含配体[分子类型]定义。
建立复合物
在pdb2gmx中,我们有一个名为“ 3HTB_processed.gro”的文件,其中包含处理过的、与力场相符的蛋白质结构。我们还从cgenff_charmm2gmx.py获得了“ jz4_ini.pdb”,它具有所有必需的H原子,并且与JZ4拓扑中的原子名称匹配。使用editconf将此.pdb文件转换为.gro格式:
gmx editconf -f jz4_ini.pdb -o jz4.gro
将3HTB_processed.gro复制到要操作的新文件中,例如,将其称为“ complex.gro”,因为在蛋白质中添加JZ4会生成我们的蛋白质-配体复合物。接下来,只需复制jz4.gro的坐标部分并将其粘贴到complex.gro中,在蛋白质原子的最后一行之下,在框矢量之前,如下所示:

建立拓扑:
在系统拓扑中包括JZ4配体的参数非常容易。在包含位置限制文件后,只需在topol.top中插入一行,显示#include“ jz4.itp”。位置限制的包含表明“蛋白质”分子类型部分的结束。

配体引入了新的二面体参数,这些参数由cgenff_charmm2gmx.py脚本写入“ jz4.prm”。在topol.top的顶部,插入#include语句以添加以下参数:

#include语句的位置很关键-它必须出现在任何[Moleculartype]条目之前,因为必须在构造任何分子之前定义所有参数。它也必须出现在父力场的#include语句之后,因为必须引入所有原子类型,然后才能引入利用它们的键合参数。
最后要做的调整是在[molecules]中。要考虑到complex.gro中存在一个新分子这一事实,我们必须在此处添加它,如下所示:

现在,拓扑和坐标文件在系统内容方面是一致的。
溶剂化
定义盒子并填充水:‘
gmx editconf -f complex.gro -o newbox.gro -bt dodecahedron -d 1.0
gmx solvate -cp newbox.gro -cs spc216.gro -p topol.top -o solv.gro
在可观察solv.gro时,您可能会想知道为什么editconf不能产生所需的十二面体晶胞形状,因为系统看起来像这样:
GROMACS程序始终使用最高效的坐标表示形式,将所有内容重新包装为三斜晶胞。mdrun执行的物理计算可以使用不同的坐标换行等效地执行,因此最有效的方法是首选。生成.tpr文件后,可以稍后恢复所需的晶胞形状。
添加离子:
用能量最小化的参数文件ions.mdp:
gmx grompp -f ions.mdp -c solv.gro -p topol.top -o ions.tpr
gmx genion -s ions.tpr -o solv_ions.gro -p topol.top -pname NA -nname CL -neutral
能量最小化:
输入参数文件为em.mdp:
gmx grompp -f em.mdp -c solv_ions.gro -p topol.top -o em.tpr
gmx mdrun -v -deffnm em
系统收敛很快。
平衡:
限制配体:
为了限制配体,我们将需要为其生成位置限制拓扑。首先,为JZ4创建一个仅包含其非氢原子的索引组:
gmx make_ndx -f jz4.gro -o index_jz4.ndx
...
> 0 & ! a H*
> q然后,执行genrestr模块并选择此新创建的索引组(在index_jz4.ndx文件中将是组3):
gmx genrestr -f jz4.gro -n index_jz4.ndx -o posre_jz4.itp -fc 1000 1000 1000
现在,我们需要将此信息包括在拓扑中。我们可以根据希望使用的条件以几种方式进行此操作。如果我们只是想在蛋白质也受到限制的情况下就限制配体,请在指定位置的拓扑中添加以下几行:
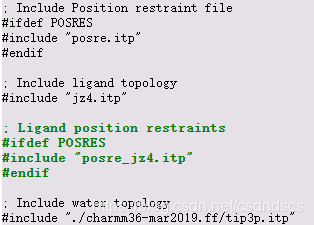
位置很重要!您必须按照指示将对posre_jz4.itp的调用放入拓扑中。jz4.itp中的参数为我们的配体定义了[分子类型]指令。分子类型以水拓扑结构(tip3p.itp)结尾。将调用放在posre_jz4.itp的任何其他位置将尝试将位置限制参数应用于错误的分子类型。
如果您希望在平衡过程中获得更多控制,即独立限制蛋白质和配体,则可以控制将配体位置限制文件包含在另一个#ifdef块中,如下所示:

在后一种情况下,要限制蛋白质和配体,我们需要在.mdp文件中指定define = -DPOSRES -DPOSRES_LIG。您要如何对待系统取决于您。这些示例仅用于说明GROMACS提供的灵活性。对于标准的平衡程序,同时限制蛋白质和配体可能就足够了。您自己的需求可能会有所不同。
温度控制
正确控制温度耦合是一个敏感的问题。将每个分子类型耦合到其自身的恒温基团是一个坏主意。例如,如果您执行以下操作:
tc-grps = Protein JZ4 SOL CL
您的系统可能会崩溃,因为温度耦合算法的稳定性不足以控制具有几个原子(即JZ4和CL)的基团产生的动能波动。不要单独耦合系统中的每个种物质。
典型的方法是设置tc-grps =Protein Non-Protein并继续。然而,“非蛋白质”组也包括JZ4。由于JZ4和蛋白质在物理上紧密相连,因此最好将它们视为单个实体。即,出于温度偶联的目的,将JZ4与蛋白质分为一组。同样,我们插入的少量Cl-被认为是溶剂的一部分。为此,我们需要一个特殊的索引组来合并蛋白质和JZ4。我们使用make_ndx模块完成此任务,提供完整系统的任何坐标文件。在这里,我使用em.gro,这是系统的输出(最小化)结构:

将“ Protein”和“ JZ4”组用下列合并,其中“>”表示make_ndx提示符:
> 1 | 13
> q现在,我们可以设置tc-grps = Protein_JZ4 Water_and_ions来实现所需的“Protein Non-Protein”效果。
用nvt.mdp文件进行NVT平衡:
gmx grompp -f nvt.mdp -c em.gro -r em.gro -p topol.top -n index.ndx -o nvt.tpr
gmx mdrun -deffnm nvt用npt.mdp进行NPT平衡:
gmx grompp -f npt.mdp -c nvt.gro -t nvt.cpt -r nvt.gro -p topol.top -n index.ndx -o npt.tpr
gmx mdrun -deffnm npt成品模拟
所用参数文件为md.mdp:
gmx grompp -f md.mdp -c npt.gro -t npt.cpt -p topol.top -n index.ndx -o md_0_10.tpr
gmx mdrun -deffnm md_0_10分析
重排坐标
就像在周期性边界条件下进行的任何模拟中一样,分子可能会突出盒子边界。将蛋白质重新置于中心并恢复菱形十二面体形状:
gmx trjconv -s md_0_10.tpr -f md_0_10.xtc -o md_0_10_center.xtc -center -pbc mol -ur compact
选择“蛋白质”作为居中,选择“系统”作为输出。请注意,对于较长的模拟(涉及跨周期性边界的多次跳跃),将复合物(蛋白质-配体,蛋白质-蛋白质)置于中心可能比较困难。在那些情况下(尤其是在蛋白质-蛋白质复合物中),可能有必要创建一个自定义索引组以用于居中,对应于复合物中一种蛋白质的活性位点或一种单体的界面残基。
用trjconv -dump提取轨迹中的第一帧(t=0s):
gmx trjconv -s md_0_10.tpr -f md_0_10_center.xtc -o start.pdb -dump 0
为了实现更加流畅的观看,执行旋转和平移拟合可能会有所帮助。执行trjconv,如下所示:
gmx trjconv -s md_0_10.tpr -f md_0_10_center.xtc -o md_0_10_fit.xtc -fit rot+trans
选择“骨架”对蛋白质骨架进行最小二乘拟合,选择“系统”进行输出。请注意,同时进行PBC重新包装和坐标拟合在数学上是不兼容的。如果您希望执行拟合(这对于可视化很有用,但对于大多数分析例程而言不是必需的),请分别执行这些坐标处理,如此处所示。
分析蛋白质-配体相互作用和配体动力学
2-丙基苯酚配体可以与Gln102侧链形成氢键。GROMACS hbond模块可轻松用于计算任何原子组之间的氢键数量,但在我们的情况下,唯一值为1或0。有关配体如何与Gln102相互作用的更详细的研究,我们将计算JZ4的羟基与Gln102的羰基O原子之间的距离。对于要形成的氢键,通常的标准是供体原子和受体原子的间距应≤3.5Å(0.35 nm)。使用距离模块,使用命令行选择语法(请参见gmx帮助选择以获取示例和更多语法),来计算轨迹上的距离。
gmx distance -s md_0_10.tpr -f md_0_10_center.xtc -select 'resname "JZ4" and name OAB plus resid 102 and name OE1' -oall
平均距离为0.31±0.05 nm,与氢键的形成一致。
通常用于确定氢键存在的第二个标准是供体,氢和受体原子之间形成的夹角。计算角度有不同的约定。在GROMACS hbond模块中,该角度定义为氢供体受体,该角度应≤30°。要执行此分析,请首先为供体原子(必须同时包括供体重原子和键合氢)和受体原子创建索引组:
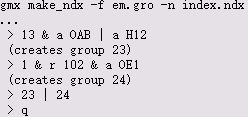
使用角度模块分析这三个原子形成的角度:
gmx angle -f md_0_10_center.xtc -n index.ndx -ov angle.xvg
请注意,与大多数其他GROMACS分析模块不同,angle不带-s参数。角度计算不需要质量或周期性信息,原子名称等,因此无需.tpr或坐标文件即可处理轨迹。该命令返回的值应约为23±9°。由于我们构建的指标组的顺序为OAB,H12,OE1,因此该结果可能有些出乎意料,这将对应于供体-氢-受体距离,我们预计该距离接近线性(〜180°)。检查索引文件的内容,您会发现:
[ JZ4_&_OAB_H12_Protein_&_r_102_&_OE1 ]
1616 2624 2636make_ndx自动将原子序数从低到高排序,因此计算的结果是受体-供体-氢角,该角与hbond模块所计算的角相同。因此,该结果与氢键的形成相一致,因为它≤30°。为了获得供体氢受体的所需角度,我们必须在文本文档里手动编辑索引组记录原子数量(2624 2636 1616)。使用该索引组重新运行角度计算得出的平均值为147±11°。
最后,我们可能对量化模拟过程中配体结合姿势发生了多少变化感兴趣。要计算仅JZ4的重原子RMSD,请为其创建一个新的索引组:
gmx make_ndx -f em.gro -n index.ndx
...
> 13 & ! a H*
> name 26 JZ4_Heavy
> q执行rms模块,选择“ Backbone”进行最小二乘拟合,选择“ JZ4_Heavy”进行RMSD计算。这样,通过拟合去除了蛋白质的整体旋转和平移,并且报告的RMSD是JZ4位置相对于蛋白质变化了多少,这很好地表明了在模拟过程中结合姿势得以保留的程度。
gmx rms -s em.tpr -f md_0_10_center.xtc -n index.ndx -tu ns -o rmsd_jz4.xvg
计算得出的RMSD应该约为0.1 nm(1Å),表明配体位置仅发生很小的变化。
蛋白质——配体相互作用能
为了量化JZ4和T4溶菌酶之间相互作用的强度,计算这两种物质之间的非键相互作用能可能是有用的。GROMACS能够分解任意数量的已定义基团之间的短距离非结合能。重要的是要注意,该数量不是自由能或结合能。实际上,大多数力场的参数化设置实际上并没有实际意义。CHARMM的参数设置为专门针对与水的量子力学相互作用能,因此它在本质上与有意义的量保持平衡,因此相互作用能非常有用。
交互能量的计算是通过.mdp文件中的energygrps关键字进行的。尽管是.mdp关键字,但不应将交互能量计算视为正常模拟的一部分。短程能量的分解与在GPU上运行不兼容,并且也不必要地减慢了计算速度。mdrun模块不需要执行额外的工作即可执行有效的模拟。因此,仅将相互作用能计算为分析的一部分,而不是动力学。从定义了energygrps = Protein JZ4的.mdp文件中创建一个新的.tpr文件,如下所示:
gmx grompp -f ie.mdp -c npt.gro -t npt.cpt -p topol.top -n index.ndx -o ie.tpr
接下来,使用-rerun选项调用mdrun以从现有的仿真轨迹重新计算能量:
gmx mdrun -deffnm ie -rerun md_0_10.xtc -nb cpu
注意,使用-deffnm读取ie.tpr并将所有输出文件写入ie。*作为其文件名。-rerun选项采用您要为其重新计算能量的轨迹的名称,而-nb cpu告诉mdrun仅尝试在CPU硬件上运行,而忽略任何可用的GPU。如上所述,这种类型的计算不能在GPU上执行。重新运行应该非常快,只需几分钟即可完成。
通过能量模块提取感兴趣的能量项。我们感兴趣的项是Coul-SR:Protein-JZ4和LJ-SR:Protein-JZ4。
gmx energy -f ie.edr -o interaction_energy.xvg
平均短程库伦相互作用能为-20.5±7.4 kJ mol-1,短程Lennard-Jones能量为-99.1±7.2 kJ mol-1。从这些量的相对大小得出结论可能很诱人,但是即使CHARMM的参数是针对显式的水相互作用能,但相互作用能进一步分解为这些成分的过程也不一定是真实的。无法通过实验验证这些数量,因此无法知道它们是否有意义。但是,在这种情况下,总的交互作用能量是有用的。该值(在按照标准公式将两个数量相加的误差传播后)为-119.6±10.3 kJ mol-1。