[For version 0.665]
CJ ([email protected])
South China Agricultural University
Overview
Rapid development of high-throughput sequencing (HTS) techniques has led biology into the “big-data” era. Data analysis using various bioinformatics softwares or pipelines relying on programming and command-line environment is challenging and time-consuming for most wet-lab biologists. Bioinformatics tools with a user-friendly interface are preferred to save time.
Thus, we present TBtools (a Toolkit for Biologists integrating various biological data handling tools), a stand-alone software with a user-friendly interface. It has powerful data handling engines for both bulk sequence processing and interactive data visualization. It includes a large collection of functions, which may facilitate much simple, routine but elaborate work on biological data, such as bulk sequence extraction, gene set enrichment analysis, Venn diagram preparation, heatmap illustration, comparative sequence visualization, etc.
A Glance of TBtools’ Functions


Download and Installation
TBtools is a platform-independent software that can be run under all operating systems with Java Runtime Environment 1.6 or newer. It is freely available to non-commercial users at
https://github.com/CJ-Chen/TBtools/releases

For users under all operating systems (Windows, Mac, Linux….):
1. Download TBtools-crossplatform_XXX.rar.
2. Unpack the rar file and obtain an executable jar file.
3. Optional. If users want to use the BLAST wrapper functions, then users is required to install BLAST package and add its bin directory to environment variables.
For users under windows, a better choice is:
1. Download TBtools_windows-32-bits-XXX.rar or TBtools_windows-64-bits-XXX.rar file.
2. Unpack the rar file and obtain an exe file.
3. Double click the exe file, click next and wait for the installation procedure.
Getting started with TBtools
For users start TBtools from a jar file, there are two ways:
1. Double click the jar file; if it don’t work, try next way.
2. Open the terminal (CMD or Powershell under Windows, Shell/Bash under Mac or Linux); type
java -Xmx2G -jar PathtoTBtools.jar
A command example under Windows
For windows users that have installed TBtools from an exe file, double click the TBtools icon and the main panel of TBtools will pop up.

In the main panel of TBtools, there is a main menubar and several buttons:
1. Click “Version” to check whether the current TBtools is the latest version.
2. Click “Citation” to get the citation method of TBtools.
Usage of Key Functions
Bulk Sequence Extraction
Go to it:
Main menubar -> Sequence Toolkits -> Fasta Tools -> Amazing Fasta Extractor
Input:
1. A target sequence file in Fasta format (ref https://en.wikipedia.org/wiki/FASTA), e.g.
>Unigene1 high expressing gene
ACGATCAGCTCAGCGACGATCGACTAGCTACGATCAGCTAGCTACGATCGACTAGCTAGCTACGA
ACGATCAGCTCAGCGACGATCGACTAGCTACGATCAGCTA
>Unigene2 low expressing gene
ACTCAGCTCAGCGACGATCAGCTCAGCGACGATCGACTAGCTACGACGACTAGCTACGA……
….
2. A set of gene identifiers or regions, e.g.
##### Lines prefixed with # will be ignored
##### Examples for One Gene ###########
Unigene_1
Unigene_2
### ChrID StartPos EndPos
Chr_1 100000 102000
### GeneID ChrID StartPos EndPos #########
FinalGeneID Chr_1 100000 120000
Output:
Complete sequences or regions of sequences specified by users
Detailed Usage:
1. Drag a target sequence file in the text-field or set it by click the “…” button
2. Click “initialize” button to build a FA-index (if the index has already been built, TBtools will skip it)
3. Set a path of an output file
4. Set a set of IDs or sequence regions
5. Click “Start”
-
Optional. If users select “Just Show Dialog”, users can obtain the extracted sequences directly from a dialog. In this case, setting of an output file is not required.

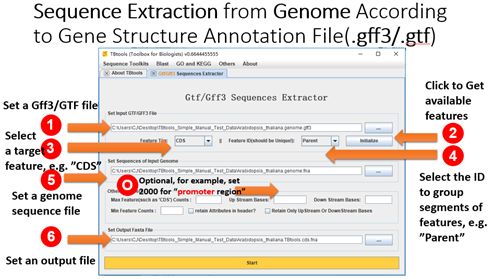
Sequence Extraction from Genome According to Gene Structure Annotation File (.gff3/gtf)
Go to it:
Main menubar -> Sequence Toolkits -> Gff3/GTF Manipulator -> Gtf/Gff3 Sequences Extractor
Input:
1. A target sequence set of genome in Fasta format
2. A corresponding gene structure annotation file in gff3/gtf format (ref https://en.wikipedia.org/wiki/General_feature_format)
Output:
A file storing sequences of a user-specific feature (CDS, exon, mRNA, gene, UTR, promoter, etc.)
Detailed Usage:
1. Set a gff3/gtf file
2. Click the “initialize” button and TBtools will provide available features for users to select
- Select a target feature, e.g. “CDS”
-
Select an ID to group sequence segments of specific features, e.g. “Parent”
5. Set a genome sequence file
6. Set an output file
*. Optional. If users want to extract sequences upstream or downstream from the specific feature, e.g 2000 bp upstream from CDS (often referring as “Promoter regions”), users need to enter “2000” in the corresponding text-field.
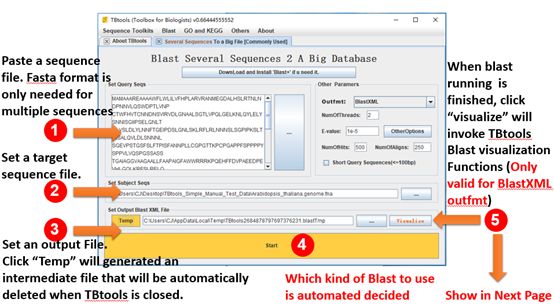
BLAST Wrapper and visualization
Go to it:
Main menubar -> Blast -> Blast Waper -> Several Sequences To a Big File [Commonly Used]
Input:
1. A set of query sequences. Fasta format is only required for multiple sequences.
2. A target sequence file, e.g. transcriptome, genome.
Output:
BLAST result in user specific format (xml, table, pairwise)
Detailed Usage:
1. Paste a set of query sequences or drag a sequence file and drop into the text-area
2. Set a target sequence file in fasta format
3. Set a path of an output file to store the BLAST result. Click “Temp” button will generate an intermediate file, which will be automatically deleted by TBtools when exited.
4. Click “Start”
5. When BLAST process is finished, user can click the “Visualize” button to invoke TBtools BLAST result visualization functions (Only valid for XML outfmt).
*. Optional. Detailed parameters could be changed on the top-right panel and the “Other Options”. Most of the time, keep it as default and it work well.
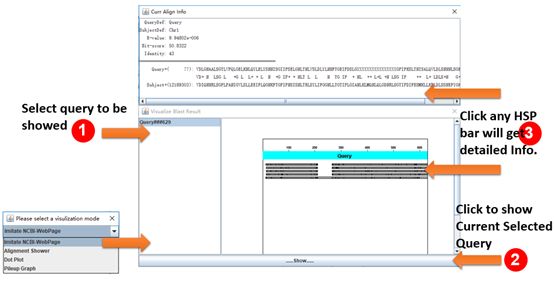
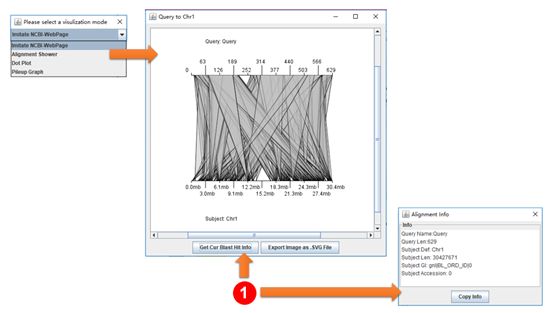
Example of visualization of BLAST results
Gene Ontology Enrichment Analysis
Go to it:
Main menubar -> GO and KEGG -> GO Enrichment
Input:
1. A go-basic.obo file downloaded from http://purl.obolibrary.org/obo/go/go-basic.obo
2. A GO annotation background, formatted as bellow. The first column are gene identifiers. If multiple gene annotated to a same GO term, comma could be used to separate them, e.g. “Unigene1,Unigene2 GO:0005509”; The second column are GO numbers. If one gene is annotated to several GO term, comma could be used to separate them, e.g. “Unigene1 GO:0008483,GO0030170”.

3. A gene set of interest for enrichment analysis.
Output:
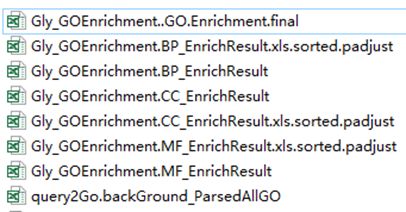
Eight files will be generated. An example is showed as bellow. GO annotation background was parsed according to Gene Ontology information stored in go-basic.obo file. Fresh-hand of enrichment analysis is recommended to use the .final.xls result directly. GO enrichment analysis is conducted to three categories, Biological Process, Cellular Component, and Molecular Function. File suffixed with “.sorted.padjust” can be used for further analyses.
Detailed Usage:
1. Set the latest go-basic.obo file
2. Set a GO annotation background
3. Set a gene set of interest
4. Set an output directory. User could also set a prefix for naming of output files.
- Optional. Click “Download go-basis.obo …” will invoke TBtools to download the latest go-basic.obo file.
-
Optional. GO-slim could be used in enrichment analysis.


KEGG Pathway Enrichment Analysis
Go to it:
Main menubar -> GO and KEGG -> KEGG Enrichment
Input:
1. A file storing KEGG pathway ontology information. Users could prepare it using the “Make One Backend File From Web” Button.
2. A KEGG annotation background, formatted as bellow. The first column are gene identifiers. If multiple gene annotated to a same KO number, comma could be used to separate them, e.g. “Unigene1,Unigene2 K12243”; The second column are KO numbers. If one gene is annotated to several KO numbers, comma could be used to separate them, e.g. “Unigene1 KO12322,KO23421”
3. A gene set of interest for enrichment analysis
Output:
Two files will be generated. *Use the .final.xls output file which has been filter with “minimum gene number>=5 and p-value<=0.05”.
Detailed Usage:
1. Set a file storing KEGG pathway ontology information.
2. Set a KEGG annotation background
3. Set a gene set of interest
4. Set an output file.
-
Optional. Select a target pathway category, e.g. “Plants”, “Animals”, ”hsa” and Click “Make …” to invoke TBtools to prepare a file storing KEGG pathway ontology information.

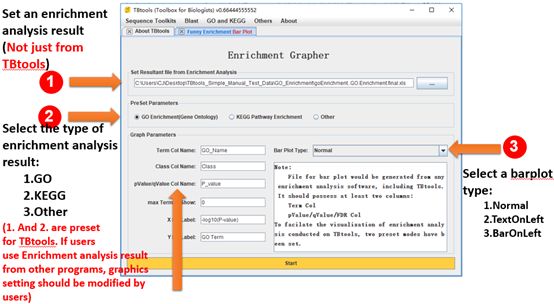
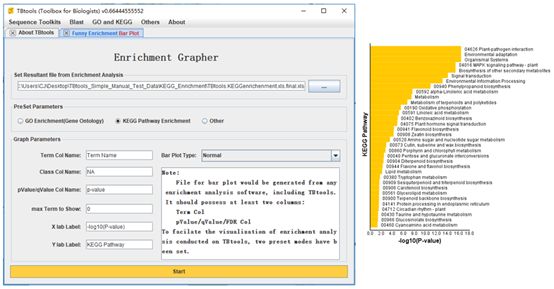
Funny Enrichment Barplot
Go to it:
Main menubar -> GO and KEGG -> Funny Enrichment Bar Plot
Input:
An enrichment analysis result. Enrichment result files from TBtools are preferred. Input file is required to contain at least two columns, i.e. a column storing “Term” labels and the other column storing “p-value”, “q-value” or “FDR”.
Output:
An interactive bar plot
Detailed Usage:
1. Set an enrichment analysis.
2. Select the type of the enrichment analysis result: “GO”, “KEGG” or “Other”. The first two options are presetting for TBtools enrichment results. If users use enrichment analysis result from other programs, Type “Other” should be selected and text-field for “Term Col Name” and “pValue….” might need to be modified.
-
Optional. Select a barplot type: “Normal”, “TextOnLeft” and “BarOnLeft”.
Example of output:

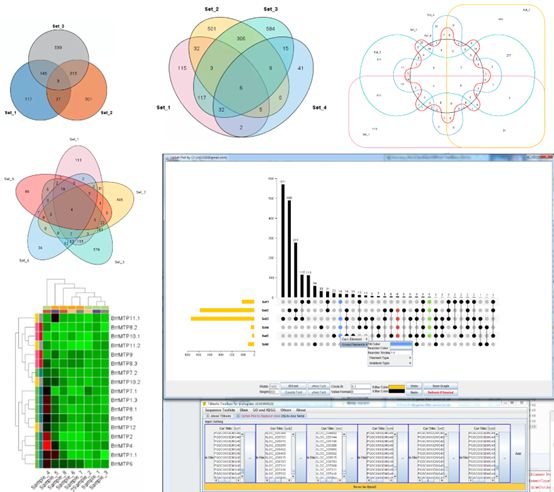
Wonderful Venn
Go to it:
Main menubar -> Graphics -> Venn and Upset Plot -> Wonderful Venn (Up to Six Sets)
Input:
Two to Six sets of identifiers
Output:
An interactive Venn diagram
Detailed Usage:
1. Paste each set of identifiers or drag and drop files storing identifiers.
2. Click “Start”
-
Optional. Change labels of each set

Example Output:


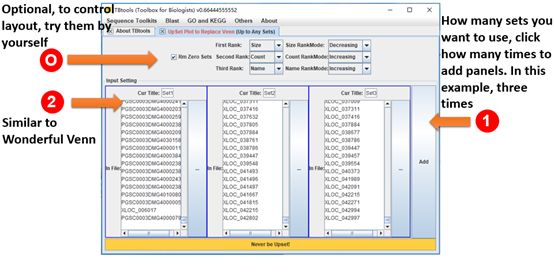
Upset Plot
Go to it:
Main menubar -> Graphics -> Venn and Upset Plot -> Upset Plot
Input:
As many sets of identifiers as users need
Output:
An interactive Upset Plot
Detailed Usage:
1. Click as many times of “Add” button to add panels as your need
2. Paste each set of identifiers or drag and drop files storing identifiers.
- Optional. Change labels of each set
- Optional. Control the rank of all vertical bar of Upset plot by changing ranking selections.
Example output:

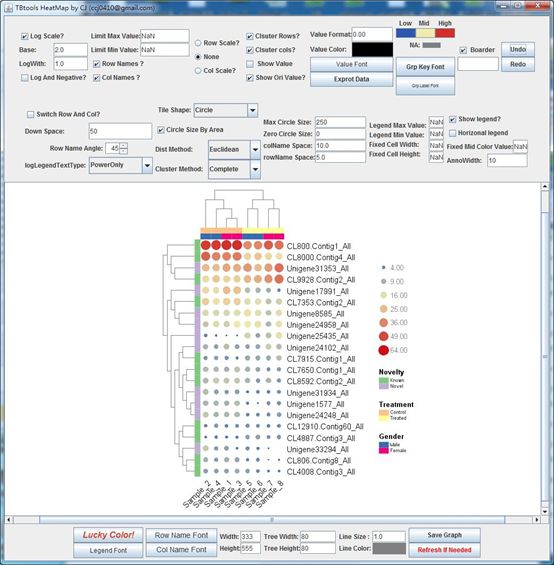
Heatmap Illustrator
Go to it:
Main menubar -> Graphics -> Amazing Heatmap -> The Amazing Simple Heatmap
Input:
1. A value matrix. The matrix should contain column names and row names, e.g.
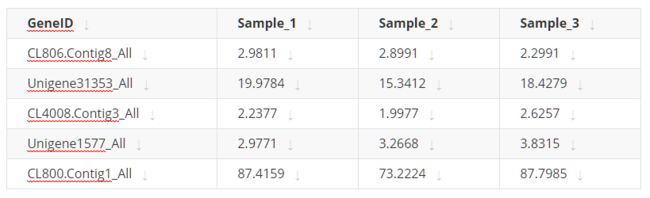
-
Optional. A tab-delimited file containing row annotation information, e.g.

- Optional. A tab-delimited file containing column annotation information, e.g.
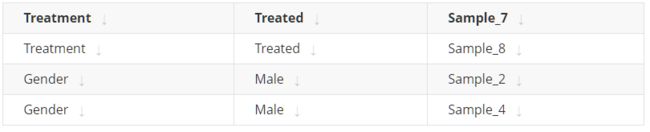
Output:
An interactive heatmap
Detailed Usage:
1. Paste the value matrix or drag and drop files storing identifiers.
2. Click “Start” - Optional. Set row annotation
- Optional. Set column annotation
- Optional. Paste a preset newick string

Example output:
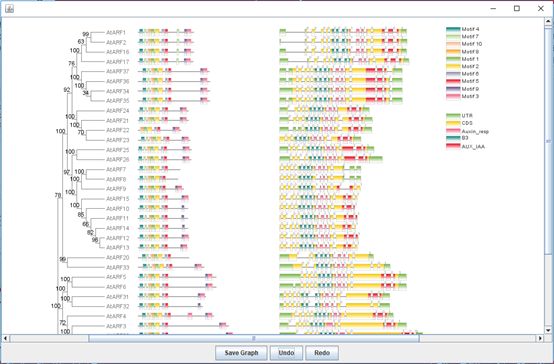
Batch Biological Sequence Visualization
Go to it:
Main menubar -> Graphics -> BioSequence Structure Illustrator -> Amazing Optional Gene Viewer
Input:
All Inputs are Optional.
A set of gene identifiers to filter the gene structure information or a newick tree string.
An XML file from MEME suite analysis result (MEME or MAST).
A gff/gtf file.
A file storing domain information in protein coordinate, e.g. analysis result from NCBI-CDD search.
A file storing domain information in mRNA coordinate, e.g. miRNA target sites.
A file storing renaming information of all sequences to be showed.
Output:
An interactive plot, which can simultaneously presenting phylogenetic tree, motif/domain patterns, gene structures, and miRNA target sites.
Detailed Usage:
All are optional.
- Optional. Paste a newick tree string or a set of gene identifiers
- Optional. Set an XML file storing motif patterns
- Optional. Set a gff/gtf file
- Optional. Set a file storing domain information in protein coordinate
- Optional. Set a file storing domain information in mRNA coordinate
- Optional. Set a file storing renaming information
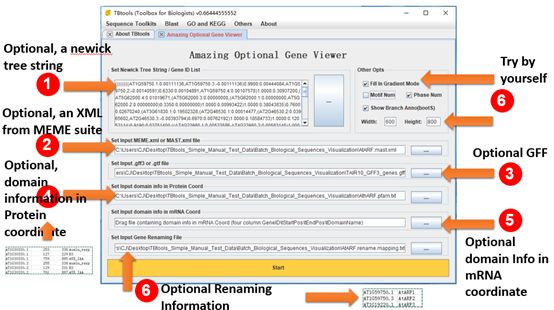
Example output: