HISAT2+STRINGTIE+BALLGOWN 分析转录组数据
师兄推荐这篇文章,按照里面的命令,先做一套转录组分析。
参考文献:
Pertea M, Kim D,Pertea G M, et al. Transcript-level expression analysis of RNA-seq experimentswith HISAT, StringTie and Ballgown.[J]. Nature Protocols, 2016, 11(9):1650.
全文链接:http://www.ccb.jhu.edu/people/infphilo/data/nprot.2016.095.pdf
我是借鉴的简书上的一篇博文,https://www.jianshu.com/p/38c2406367d5,谢谢这个博主啦!
数据 :https://pan.baidu.com/s/1aX93Q65Dit3iqslRWkQcsw
genes 针对基因组的注释文件.gtf
genome 染色体X的序列文件 chrX.fa
geuvadis_phenodata.csv
mergelist.txt 以上两个都是之前博主创建表明数据关系的文件
indexes hisat2对于染色体X的indexes文件,1~8.ht2 索引文件
samples 数据 fastq.gz
文章背景: 见文章
~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~
转录组分析 背景知识:
从原始RNA-Seq数据着手,质控——建立索引文件——比对、拼接、排序——初组装——合并——计算表达量,并输出为baoogown格式——进行差异分析——作图,这里输出结果包括基因list、转录本,及每个样本的表达量,能表现差异表达基因的表格 并完成显著性计算。
在R里使用ballgown处理需要得到: #了解一下就OK
1. 表型数据 关于样本的信息
2. 表达数据 标准化和未标准化的关于外显子,junction,转录本,基因的表达数量
3. 基因信息 有关外显子,junction,转录本,基因的坐标以及注释信息
大多数差异表达分析都会包括一下几个步骤: #需要着重理解
1. 数据可视化和检查
2. 差异表达的统计分析
3. 多重检验校正
4. 下游检查和数据summary
ballgown的使用: #分析过程的难点在ballgown,提前理解有助于后面,现在回过头来看还是很懵逼……
1. 数据的读入
2. 预测丰度的检查:以FPKM为单位的丰度预测将会根据library size进行标准化。FPKM(fragments per kilobase of transcript per million mapped reads)
3. 使用线性模型进行差异表达分析,由于FPKM对于转录本解读过于曲解,所以这里需要使用log转化处理数据,随后再使用线性模型进行差异分析。
4. ballgown可以对于time-course和fixed_conduction数据进行差异分析,但是无法避免批次效应带来的误差。# 使用stattest功能可以指定任何已知的confounder
5. ballgown 可以帮助你在基因、转录本、外显子、junction水平上进行差异分析。
6. 结果会以表格形式展出,如果样本多还有p值和q值。
7. 结果数据可以用来进行下一步的gene set分析(例如GSEA)等等。
~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~
正式分析开始(提前把文件解压好,数据解压出来&提前装好hisat2,stringtie,samtools,R, ballgown到后面再装)
一、 质控,运行fastQC
nohup fastqc filename.fq &
for i in *fastq;
do
i=${i%fastq*};
fastqc ${i}fastq
done
#这是一个循环语句,文件12个,一个一个来就很累了,但是我也没有理解这个循环语句,照葫芦画瓢而已,改一下路径
#输出文件为filename.fastqc.zip,可在网页上看结果,或者Xmanager 6查看,因为文章里的数据很好,不用再进行筛选,就直接下一步 PS, nohup & 语句,是使命令在后台运行。
二、 HISAT2
1. 使用hisat2-build建立索引文件(我的文件里已有索引文件,就不需要这一步)
hisat2-build -f /注释文件路径/genome.fa /索引文件输出路径/文件名.X.ht2
# -f 指定注释文件格式,默认为fa,可不填 运行成功后,会产生如下文件name.1.ht2 …… name.8.th2
2. 使用生成的索引文件对RNA-Seq进行比对拼接
nohup hisat2 -p 8-x /home/czou/lixin/b/chrX_data/indexes/ -1 ERR188044_chrX_1.fastq -2 ERR188044_chrX_2.fastq -S ERR188044.sam &
for i in *1.fastq;
do
i=${i%1.fastq*};
hisat2 -p 8-x /home/czou/lixin/b/chrX_data/indexes/chrX_tran -1 ${i}1.fastq -2 ${i}2.fastq -S ${i}.sam
done
3. SAM tools 排序及格式转换
samtools sort -@ 8 -o ERR188044_chrX_.bam ERR188044_chrX_.sam
for i in *.sam;
do
i=${i%.sam*};
samtools sort -@ 8 -o ${i}.bam ${i}.sam
done
三、 stringtie 序列初组装
stringtie -p 8 -G /home/czou/lixin/b/chrX_data/genes/chrX.gtf -o ERR188044_chrX_.gtf ERR188044_chrX_.bam
for i in *.bam;
do
i=${i%.bam*};
nohup stringtie -p 8 -G /home/czou/lixin/b/chrX_data/genes/chrX.gtf -o ${i}.gtf ${i}.bam &
done
合并
stringtie --merge -p 8 -G /home/czou/lixin/b/chrX_data/genes/chrX.gtf -o stringtie_merged.gtf /home/czou/lixin/b/chrX_data/mergelist.txt
四、 检测转录本与参考注释的比较
gffcompare -r /home/czou/lixin/b/chrX_data/genes/chrX.gtf -G -o merged stringtie_merged.gtf
评估表达量,并为ballgown包提供输入文件
stringtie -e -B -p 8 -G /home/czou/lixin/b/chrX_data/samples/sam/gtf2/stringtie_merged.gtf -o ballgown/ERR188044_chrX/ERR188044_chrX.gtf /home/czou/lixin/b/chrX_data/samples/fastq/ERR188044_chrX_.bam
for i in *_chrX_.bam;
do
i=${i%_chrX_.bam*}; stringtie -e -B -p 8 -G /home/czou/lixin/b/chrX_data/samples/sam/gtf2/stringtie_merged.gtf -o ballgown/${i}/${i}_chrX.gtf /home/czou/lixin/b/chrX_data/samples/fastq/${i}_chrX_.bam
done
~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~
Ballgown包
RSkittleBrewer(用于设置颜色)
genefilter(用于快速计算均值和方差)
dplyr(用于分类和排列结果)
devtools(用于再现性和安装包)
~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~
五、 差异表达分析(在R中进行)
1. 安装
source("https://bioconductor.org/biocLite.R")
biocLite("ballgown")
library(dplyr)
install.packages("dplyr")
library(devtools)
install.packages("devtools")
library(genefilter)
source("https://bioconductor.org/biocLite.R")
biocLite("genefilter")
library("RSkittleBrewer")
devtools::install_github('RSkittleBrewer', 'alyssafrazee')
#加载要用的语言包
library(ballgown)
library(RSkittleBrewer)
library(genefilter)
library(dplyr)
library(devtools)
getwd()
set() # 修改当前路径至数据位置,加“”
read.csv("geuvadis_phenodata1.csv") #读取表型数据,数据分组信息
pheno_data <- read.csv("geuvadis_phenodata1.csv")
bg_chrX = ballgown(dataDir = "C:/Users/Administrator/Documents/R/hisat2 stringtie ballgown/ballgown", samplePattern = "ERR", pData = pheno_data)
#dataDir 告知数据路径, samplePattern 依据样本的名字来, pData 指明数据关系,这里面的第一列样本必须和ballgown下面的文件名一样,否则会报错。
bg_chrX_filt = subset(bg_chrX, "rowVars(texpr(bg_chrX)) > 1", genomesubset = TRUE)
#滤掉低丰度的基因,这里选择过滤掉样本间差异少于一个转录本的数据
result_transcripts = stattest(bg_chrX_filt, feature = "transcript", covariate = "sex", adjustvars = c("population"), getFC = TRUE, meas = "FPKM")
result_transcripts #确定组件有差异的转录本,在这里我们比较的性别之间的基因差异,指定的分析参数是“transcripts”,主变量是“sex",修正变量是
”population",getFC可以指定输出结果显示组间表达量的foldchange。
result_genes = stattest(bg_chrX_filt, feature = "gene", covariate = "sex", adjustvars = c("population"), getFC = TRUE, meas = "FPKM")
result_genes #确定各组间有差异的基因
result_transcripts = data.frame(geneNames = ballgown::geneNames(bg_chrX_filt), geneIDs = ballgown::geneIDs(bg_chrX_filt), result_transcripts)
result_transcripts #为组间有差异的转录本添加基因名?????不理解
result_transcripts = arrange(result_transcripts, pval) #按照p-value值从小到大排序
result_genes = arrange(result_genes, pval)
write.csv(result_transcripts, "chrX_transcript_results.csv", row.names = FALSE) #将两个结果写到csv文件中
write.csv(result_genes, "chrX_gene_results.csv", row.names = FALSE)
subset(result_transcripts, result_transcripts$qval < 0.05) #从以上的输出中选出q值<0.05的转录本和基因,即性别表达有差异的基因
subset(result_genes, result_genes$qval < 0.05)
tropical = c('darkorange', 'dodgerblue', 'hotpink', 'limegreen', 'yellow') #赋予调色板5个指定颜色
palette(tropical)
fpkm = texpr(bg_chrX, meas = "FPKM") #以FPKM为参考值作图,以性别作为区分条件
fpkm = log2(fpkm + 1) #方便作图,将其log转化,+1是为了别面出现log2(0)的情况
boxplot(fpkm, col = as.numeric(pheno_data$sex), las = 2, ylab = 'log2(FPKM + 1)')
#以上完整做出箱线图
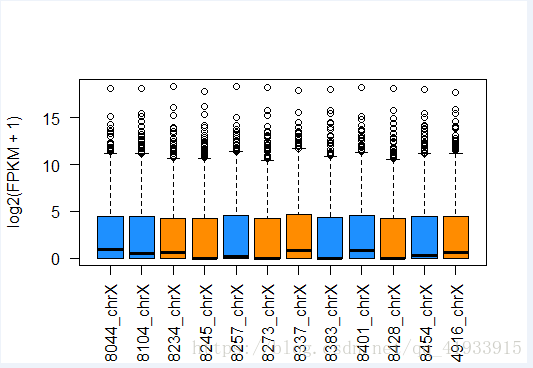
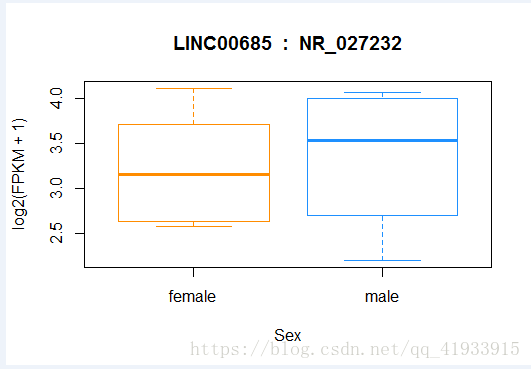
#就单个转录本做箱线图
ballgown::transcriptNames(bg_chrX)[12]
ballgown::geneNames(bg_chrX)[12]
plot(fpkm[12,] ~ pheno_data$sex, border = c(1,2),
main = paste(ballgown::geneNames(bg_chrX)[12],' : ',
ballgown::transcriptNames(bg_chrX)[12]), pch = 19, xlab = "Sex",
ylab = 'log2(FPKM + 1)')
points(fpkm[12,] ~ jitter(as.numeric(pheno_data$sex)),
col = as.numeric(pheno_data$sex)) # 这一步作用是??male做出来和原图有细微不同
#查看某一基因位置上所有转录本
# plotTranscripts函数可以根据指定基因的id画出在特定区段的转录本
# 可以通过sample函数指定看在某个样本中的表达情况,这里选用id=1750, sample=ERR188234
plotTranscripts(ballgown::geneIDs(bg_chrX)[1750], bg_chrX, main = c('Gene MSTRG.575 in sample ERR188234'), sample = c('ERR188234'))
#不成功???
#以性别为区分,查看表达情况
# 这里以id=575的基因为例(对应上一步作图)
plotMeans('MSTRG.575', bg_chrX_filt, groupvar = "sex", legend = FALSE)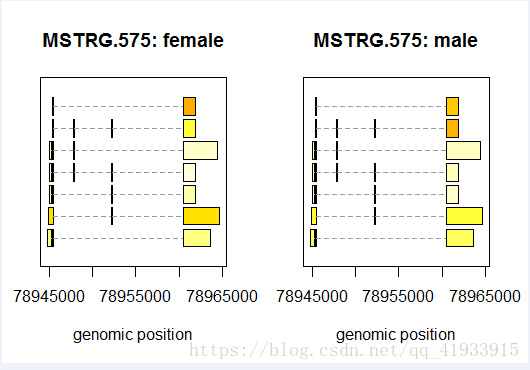
还有一个图没有做成功,慢慢去找原因吧,ganbade