单细胞转录组(scRNA-seq)分析01 | Scater包的使用
文章目录
- 一、介绍
- 二、工作流
- 三、常用函数
- plotColData
- plotExplanatoryVariables
- plotExpression
- plotExprsFreqVsMean
- plotExprsVsTxLength
- plotHeatmap
- plotHighestExprs
- plotPlatePosition
- plotQC
- plotReducedDim
- plotRLE
- plotRowData
- plotScater
- Reduced dimension plots
一、介绍
- 用于单细胞RNA-seq数据
- 提供严格的质量控制:将原始测序读数处理为可用于下游分析的高质量表达数据集
- 提供了丰富的绘图工具套件
- R包地址:http://bioconductor.org/packages/scater
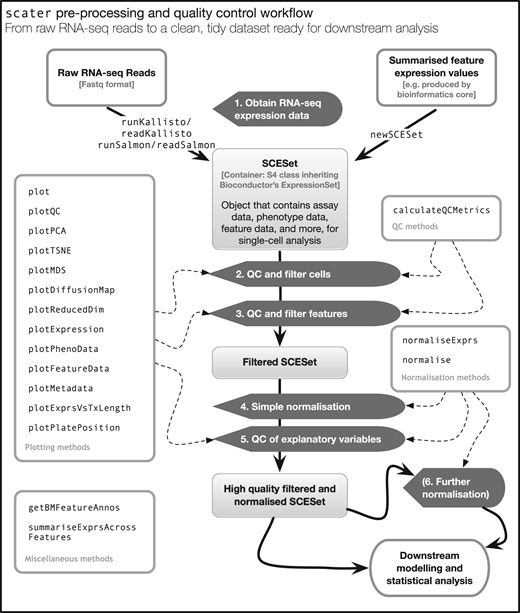
二、工作流
三、常用函数
plotColData
# 导入包
suppressMessages(library(scater))
suppressMessages(library(scRNAseq))
# 载入示例数据
data("sc_example_counts")
data("sc_example_cell_info")
# 构建 SingleCellExperiment 对象
example_sce <- SingleCellExperiment(
assays = list(counts = sc_example_counts),
colData = sc_example_cell_info
)
# 计算 SingleCellExperiment 对象中每个特征和细胞的质控标准
example_sce <- calculateQCMetrics(example_sce)
# 计算 SingleCellExperiment 对象中read计数矩阵的归一化表达值
example_sce <- normalize(example_sce)
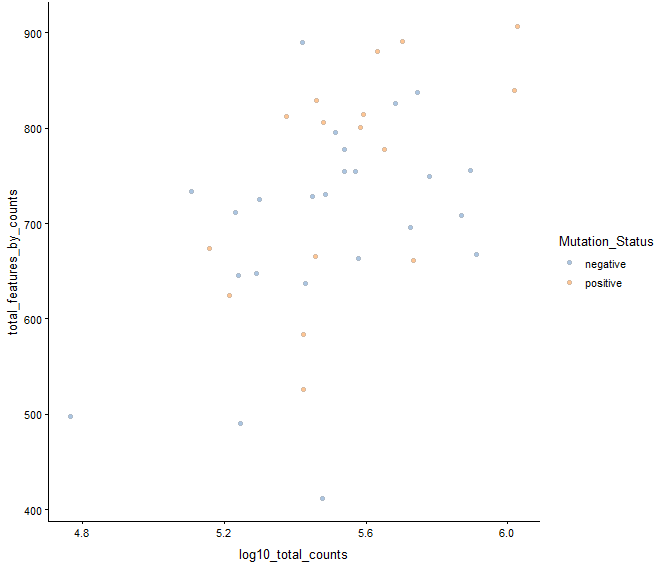
plotColData(example_sce, y = "total_features_by_counts",
x = "log10_total_counts", colour_by = "Mutation_Status")
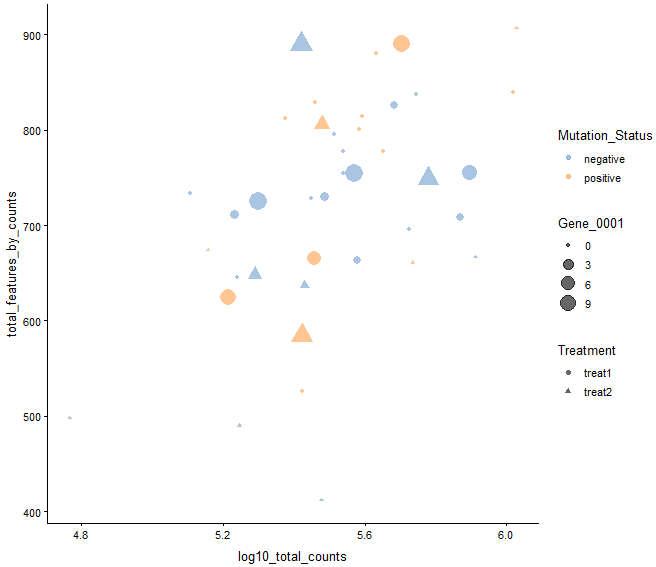
plotColData(example_sce, y = "total_features_by_counts",
x = "log10_total_counts", colour_by = "Mutation_Status",
size_by = "Gene_0001", shape_by = "Treatment")
plotColData(example_sce, y = "Treatment",
x = "log10_total_counts", colour_by = "Mutation_Status")
plotColData(example_sce, y = "total_features_by_counts",
x = "Cell_Cycle", colour_by = "Mutation_Status")
plotExplanatoryVariables
解释变量(ExplanatoryVariables):https://www.statisticshowto.datasciencecentral.com/explanatory-variable/
解释变量是一种自变量。这两个术语通常可互换使用。但是,是两者之间的细微差别。当一个变量是独立的,它不影响在所有的任何其他变量。当变量不是独立的时候,它是一个解释变量。 它在临床研究中非常重要。对于大多数情况,特别是在统计数据中,这两个术语基本相同。
假设您有两个变量来解释体重增加:快餐和苏打水。虽然你可能认为吃快餐和喝苏打水是相互独立的,但它们并不是真的。那是因为快餐店鼓励你在用餐时买苏打水。如果你停在某个地方买苏打水,那里经常会有很多快餐选择,比如热狗。虽然这些变量并非完全相互独立,但它们确实会对体重增加产生影响。它们被称为解释变量,因为它们可能为体重增加提供一些解释。
data("sc_example_counts")
data("sc_example_cell_info")
example_sce <- SingleCellExperiment(
assays = list(counts = sc_example_counts),
colData = sc_example_cell_info
)
example_sce <- normalize(example_sce)
plotExplanatoryVariables(example_sce)
plotExpression
data("sc_example_counts")
data("sc_example_cell_info")
example_sce <- SingleCellExperiment(
assays = list(counts = sc_example_counts),
colData = sc_example_cell_info
)
example_sce <- calculateQCMetrics(example_sce)
sizeFactors(example_sce) <- colSums(counts(example_sce))
example_sce <- normalize(example_sce)
# 前十五个基因的表达值
plotExpression(example_sce, 1:15)
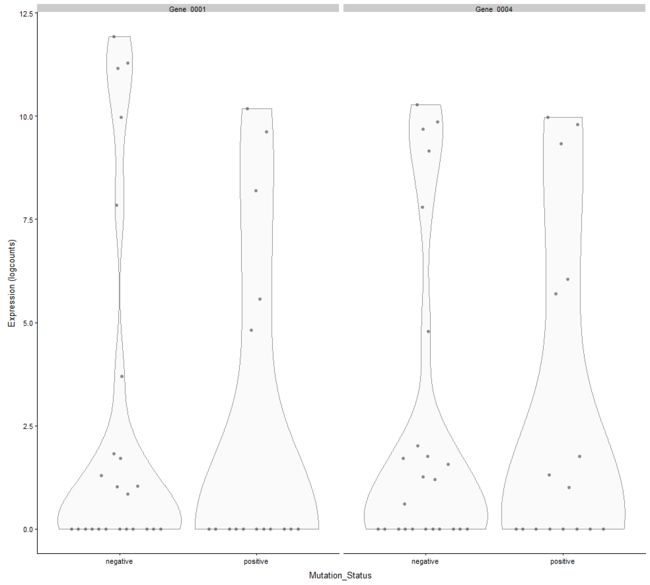
plotExpression(example_sce, c("Gene_0001", "Gene_0004"), x="Mutation_Status")
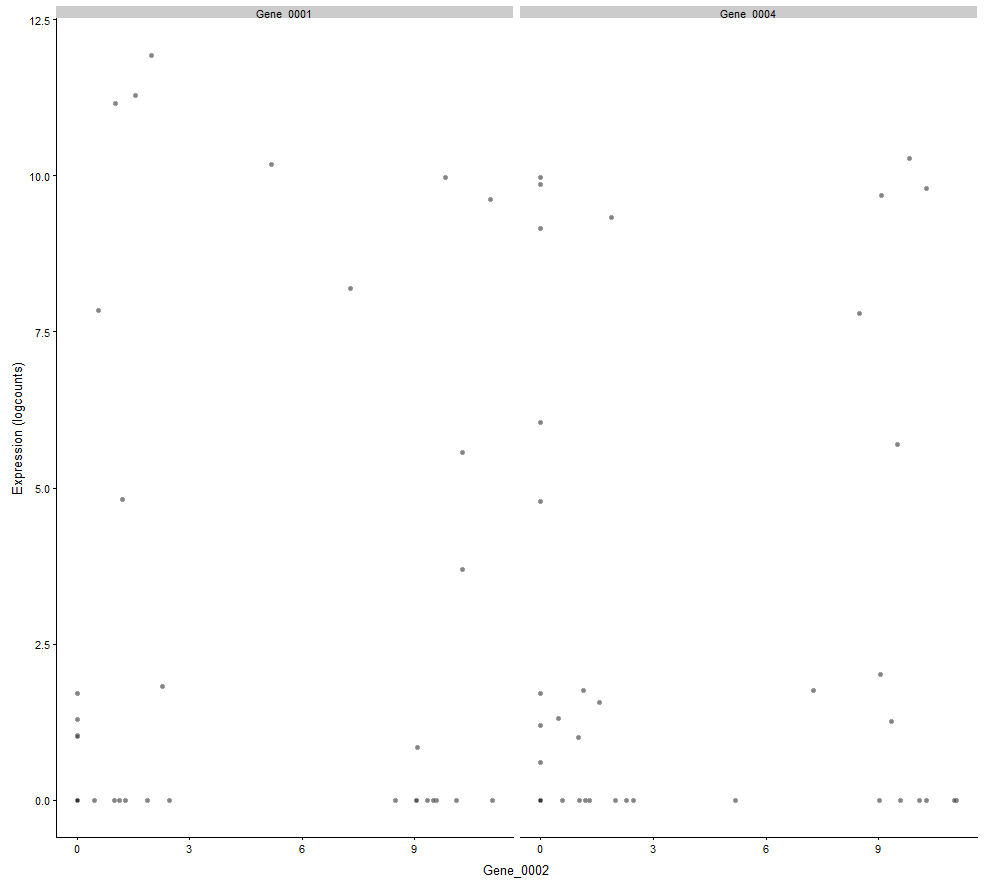
plotExpression(example_sce, c("Gene_0001", "Gene_0004"), x="Gene_0002")
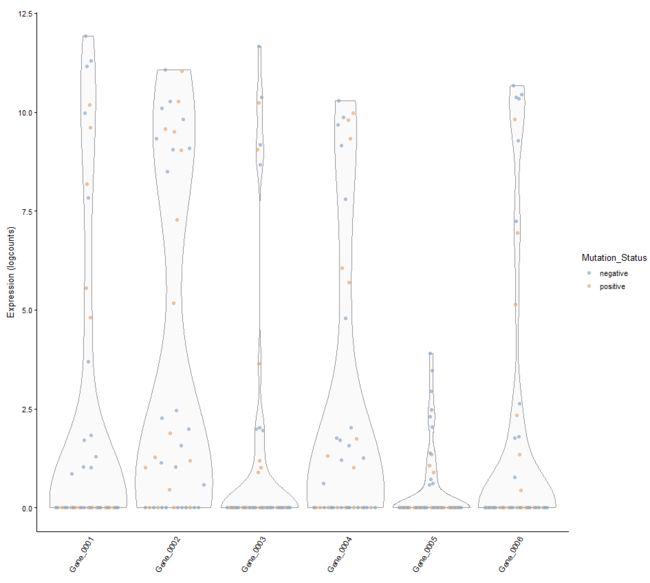
plotExpression(example_sce, 1:6, colour_by = "Mutation_Status")
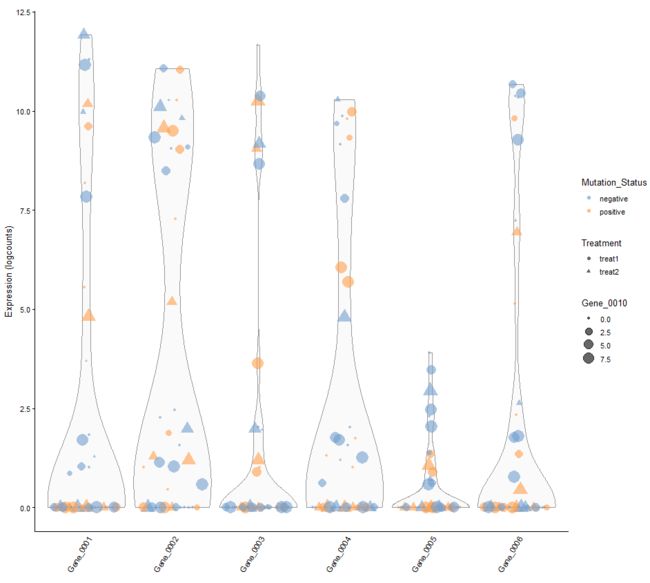
plotExpression(example_sce, 1:6, colour_by = "Mutation_Status",
shape_by = "Treatment", size_by = "Gene_0010")
plotExpression(example_sce, 1:4, "Gene_0004", show_smooth = TRUE)
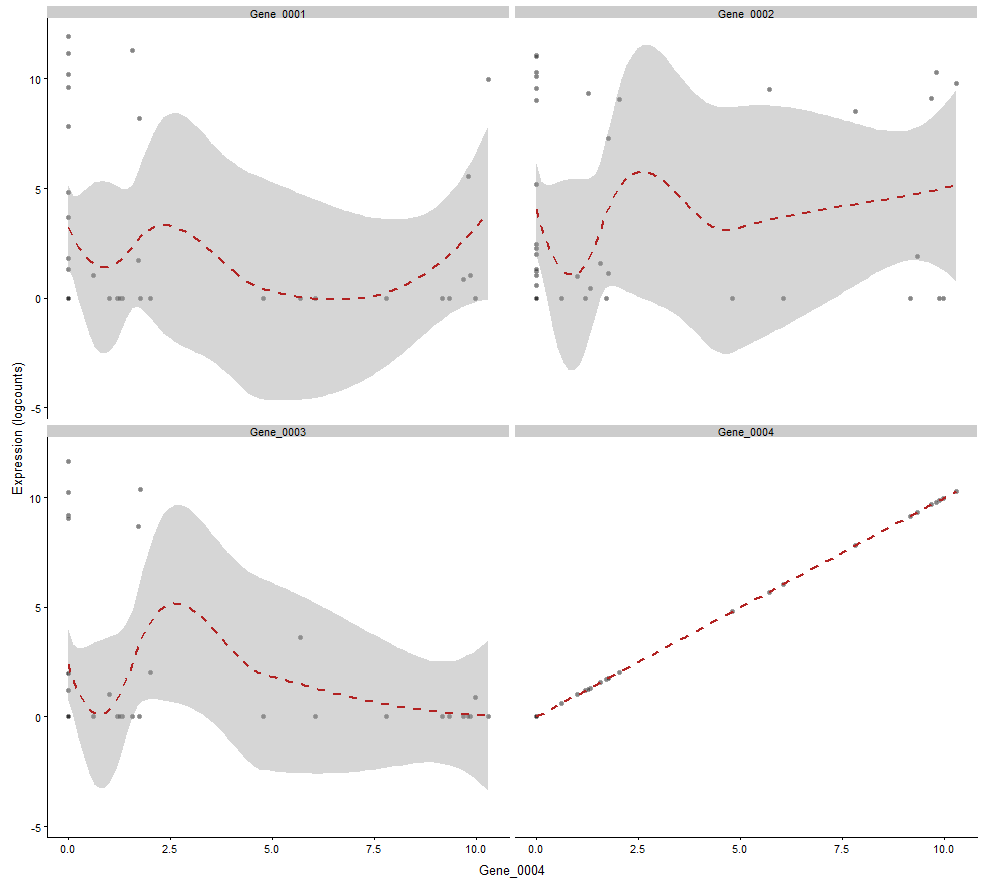
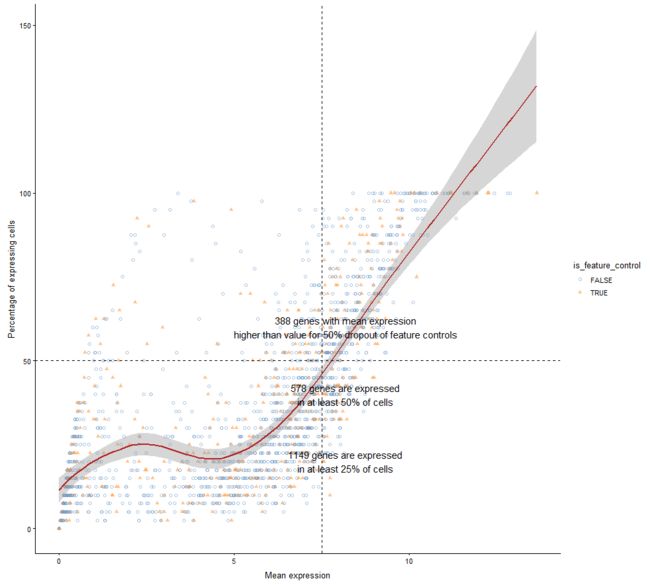
plotExprsFreqVsMean
表达频率(即表达细胞的百分比)Vs SingleCellExperiment对象中每个特征的平均表达水平
data("sc_example_counts")
data("sc_example_cell_info")
example_sce <- SingleCellExperiment(
assays = list(counts = sc_example_counts),
colData = sc_example_cell_info
)
example_sce <- normalize(example_sce)
example_sce <- calculateQCMetrics(example_sce,
feature_controls = list(set1 = 1:500))
plotExprsFreqVsMean(example_sce)
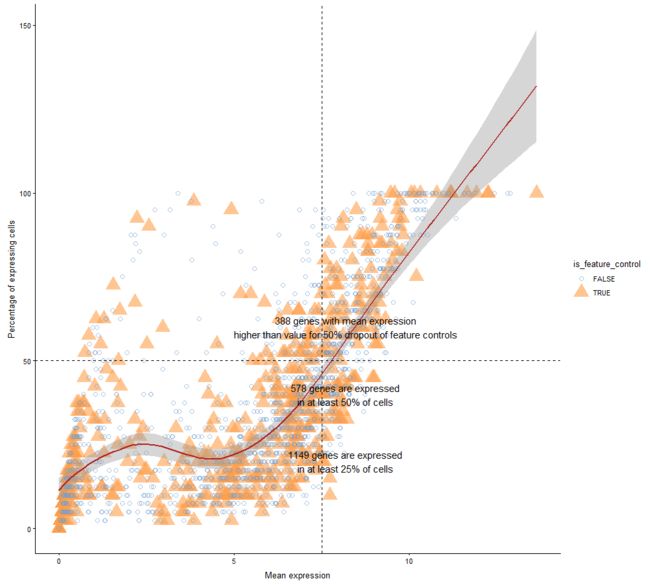
plotExprsFreqVsMean(example_sce, size_by = "is_feature_control")
plotExprsVsTxLength
Plot mean expression values for all features in a SingleCellExperiment object against transcript length values.
data("sc_example_counts")
data("sc_example_cell_info")
rd <- DataFrame(gene_id = rownames(sc_example_counts),
feature_id = paste("feature", rep(1:500, each = 4), sep = "_"),
median_tx_length = rnorm(2000, mean = 5000, sd = 500),
other = sample(LETTERS, 2000, replace = TRUE)
)
rownames(rd) <- rownames(sc_example_counts)
example_sce <- SingleCellExperiment(
assays = list(counts = sc_example_counts),
colData = sc_example_cell_info, rowData = rd
)
example_sce <- normalize(example_sce)
plotExprsVsTxLength(example_sce, "median_tx_length")
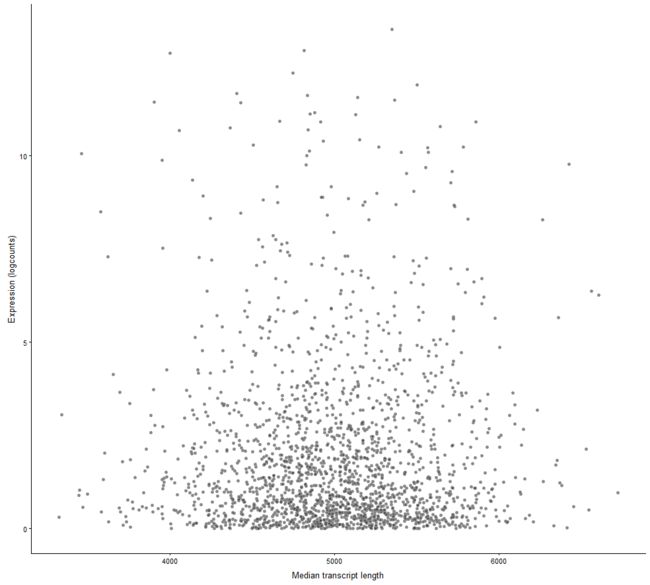
plotExprsVsTxLength(example_sce, "median_tx_length", show_smooth = TRUE)
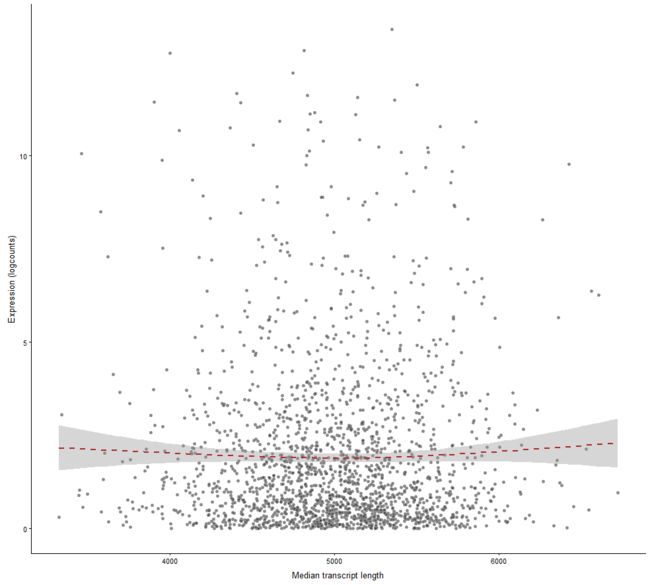
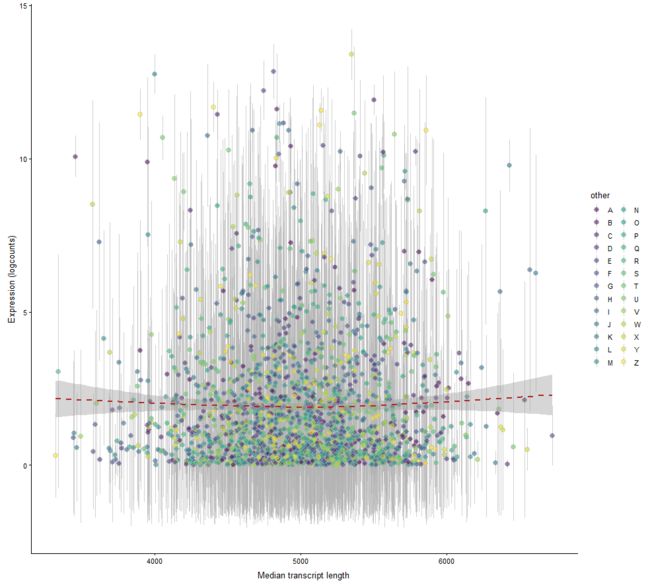
plotExprsVsTxLength(example_sce, "median_tx_length", show_smooth = TRUE,
colour_by = "other", show_exprs_sd = TRUE)
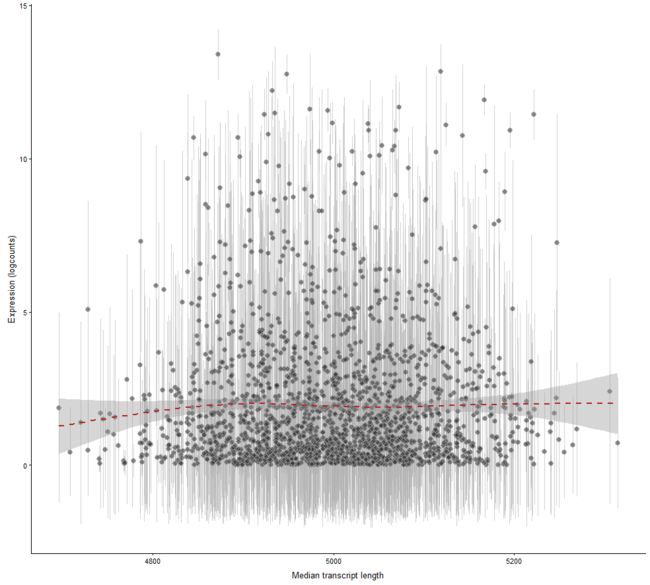
## using matrix of tx length values in assays(object)
mat <- matrix(rnorm(ncol(example_sce) * nrow(example_sce), mean = 5000,
sd = 500), nrow = nrow(example_sce))
dimnames(mat) <- dimnames(example_sce)
assay(example_sce, "tx_len") <- mat
plotExprsVsTxLength(example_sce, "tx_len", show_smooth = TRUE,
length_is_assay = TRUE, show_exprs_sd = TRUE)
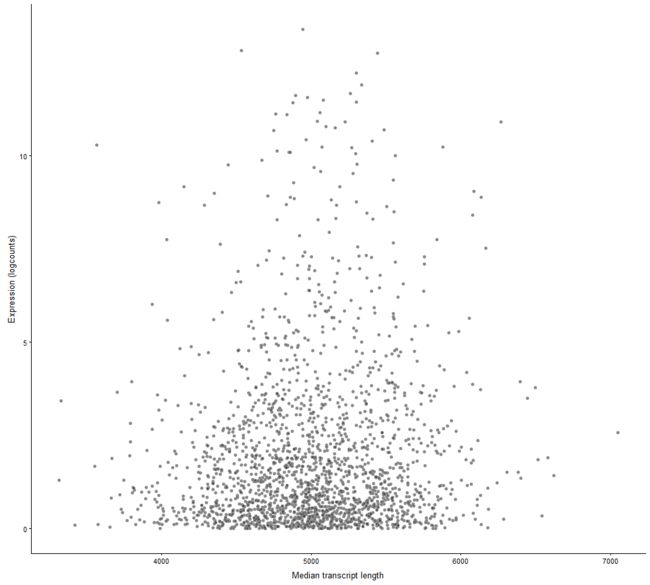
## using a vector of tx length values
plotExprsVsTxLength(example_sce,
data.frame(rnorm(2000, mean = 5000, sd = 500)))
plotHeatmap
Create a heatmap of expression values for each cell and specified features in a SingleCellExperiment
object.
example(normalizeSCE) # borrowing the example objects in here.
plotHeatmap(example_sce, features=rownames(example_sce)[1:10])
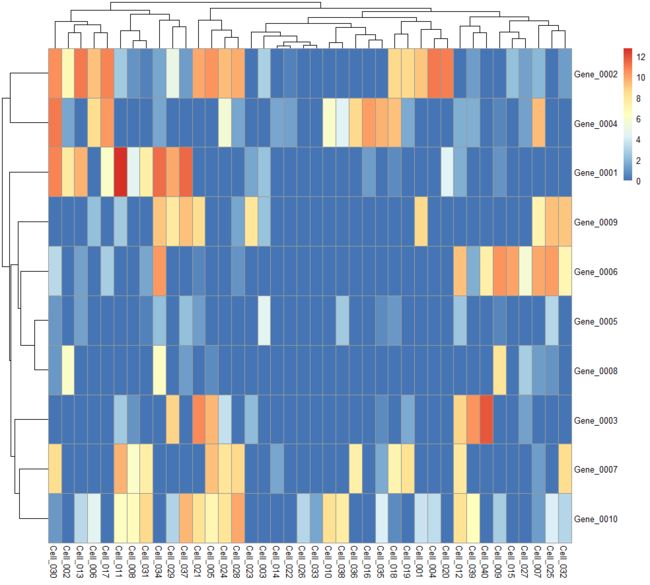
plotHeatmap(example_sce, features=rownames(example_sce)[1:10],
center=TRUE, symmetric=TRUE)
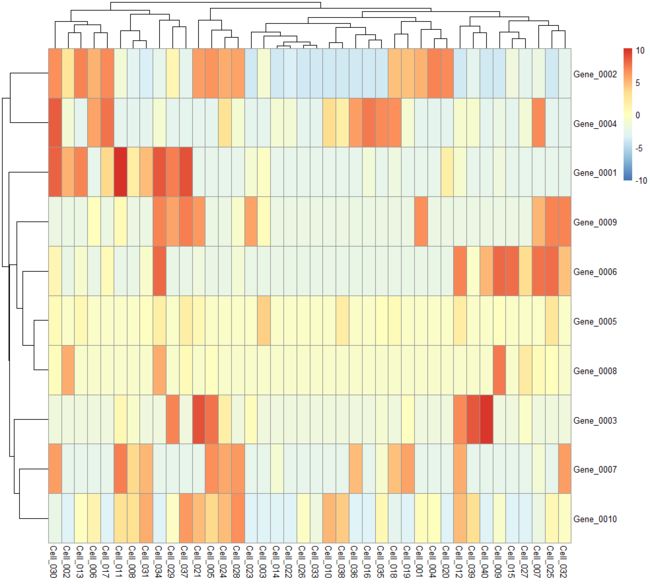
plotHeatmap(example_sce, features=rownames(example_sce)[1:10],
colour_columns_by=c("Mutation_Status", "Cell_Cycle"))

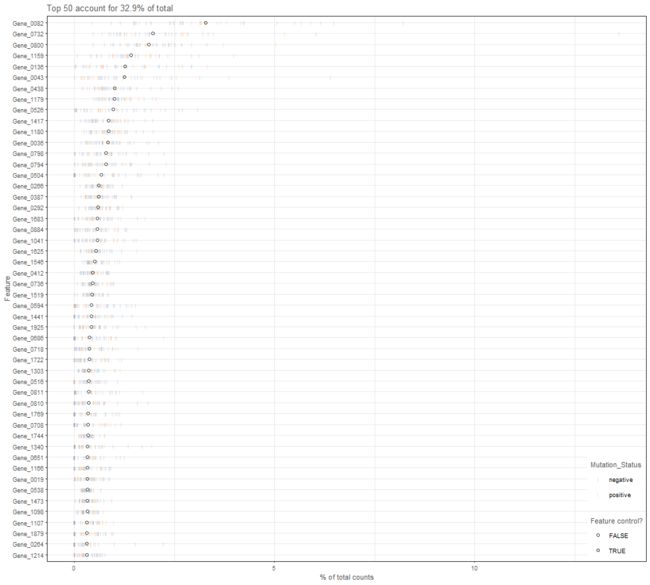
plotHighestExprs
Plot the features with the highest average expression across all cells, along with their expression in
each individual cell.
data("sc_example_counts")
data("sc_example_cell_info")
example_sce <- SingleCellExperiment(
assays = list(counts = sc_example_counts),
colData = sc_example_cell_info
)
example_sce <- calculateQCMetrics(example_sce,
feature_controls = list(set1 = 1:500)
)
plotHighestExprs(example_sce, colour_cells_by ="total_features_by_counts")
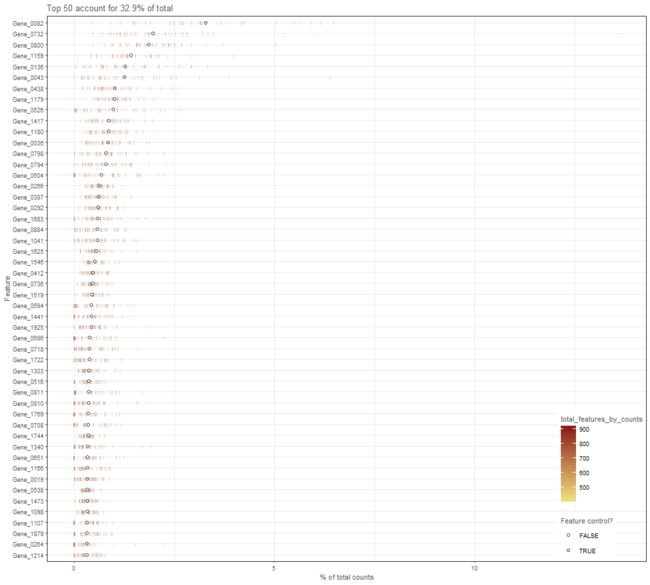
plotHighestExprs(example_sce, controls = NULL)
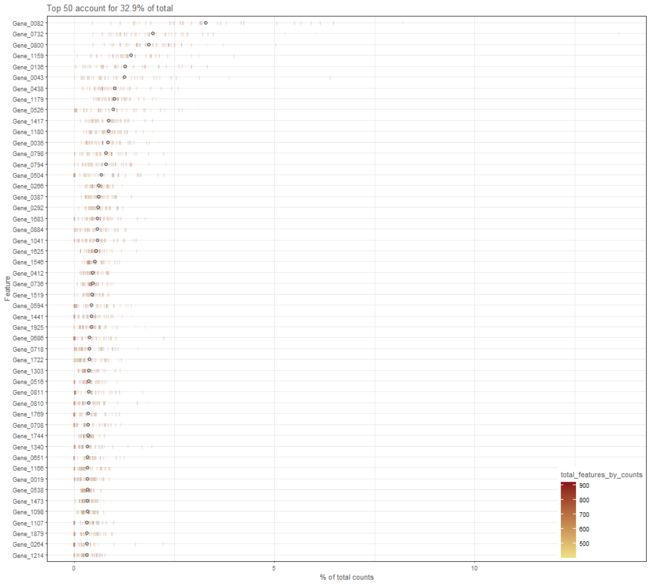
plotHighestExprs(example_sce, colour_cells_by="Mutation_Status")
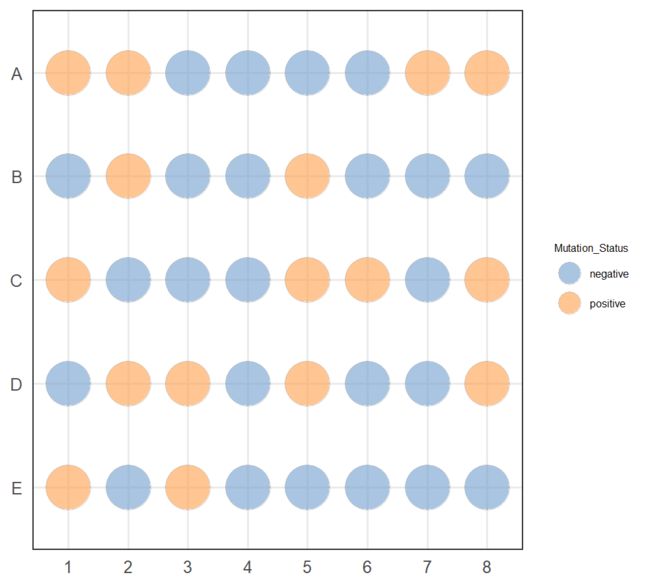
plotPlatePosition
Plots cells in their position on a plate, coloured by metadata variables or feature expression values
from a SingleCellExperiment object.
## prepare data
data("sc_example_counts")
data("sc_example_cell_info")
example_sce <- SingleCellExperiment(
assays = list(counts = sc_example_counts),
colData = sc_example_cell_info
)
example_sce <- normalize(example_sce)
example_sce <- calculateQCMetrics(example_sce)
## define plate positions
example_sce$plate_position <- paste0(
rep(LETTERS[1:5], each = 8),
rep(formatC(1:8, width = 2, flag = "0"), 5)
)
## plot plate positions
plotPlatePosition(example_sce, colour_by = "Mutation_Status")
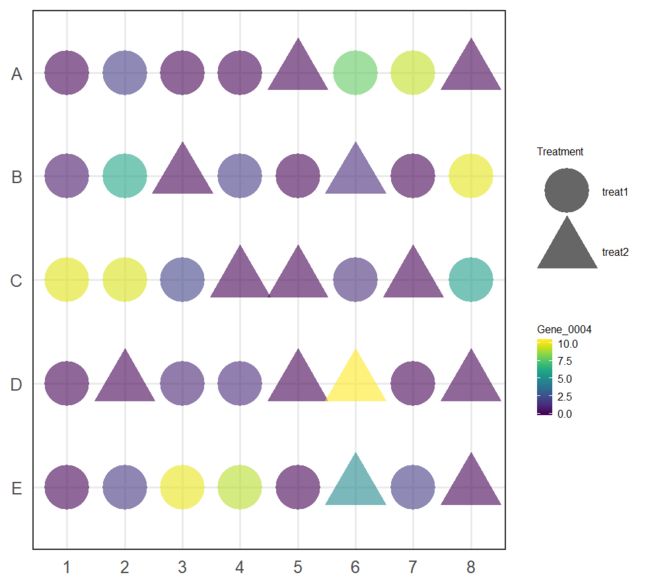
plotPlatePosition(example_sce, shape_by = "Treatment", colour_by = "Gene_0004")
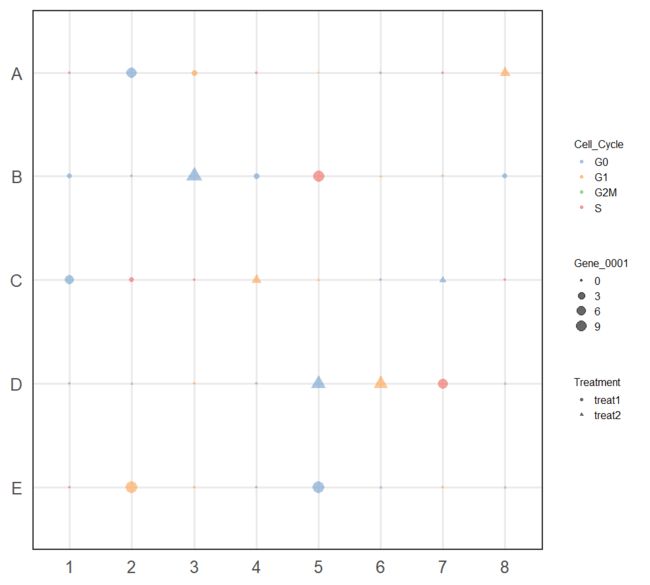
plotPlatePosition(example_sce, shape_by = "Treatment", size_by = "Gene_0001",
colour_by = "Cell_Cycle")
plotQC
Produce QC diagnostic plots
data("sc_example_counts")
data("sc_example_cell_info")
example_sce <- SingleCellExperiment(
assays = list(counts = sc_example_counts),
colData = sc_example_cell_info)
example_sce <- normalize(example_sce)
example_sce <- calculateQCMetrics(example_sce)
plotQC(example_sce, type="high", colour_cells_by="Mutation_Status")
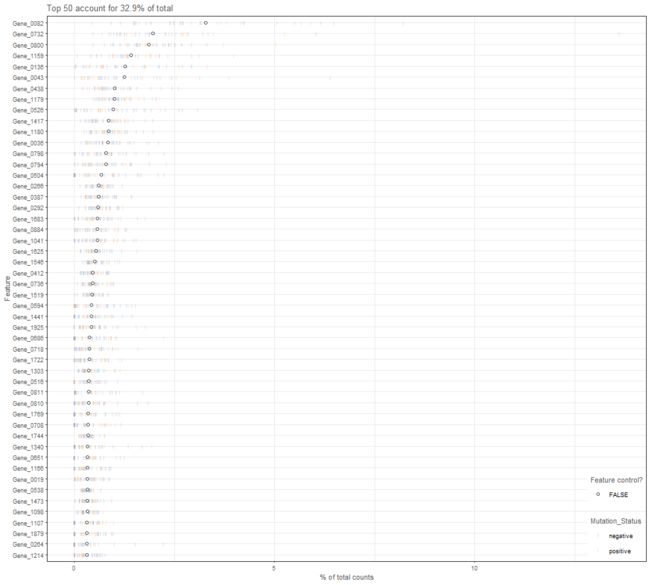
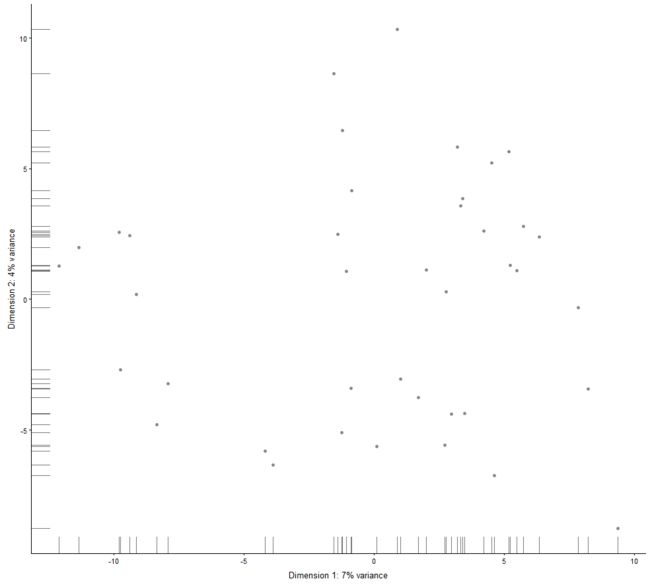
plotReducedDim
Plot cell-level reduced dimension results stored in a SingleCellExperiment object.
data("sc_example_counts")
data("sc_example_cell_info")
example_sce <- SingleCellExperiment(
assays = list(counts = sc_example_counts),
colData = sc_example_cell_info
)
example_sce <- normalize(example_sce)
example_sce <- runPCA(example_sce, ncomponents=5)
plotReducedDim(example_sce, "PCA")
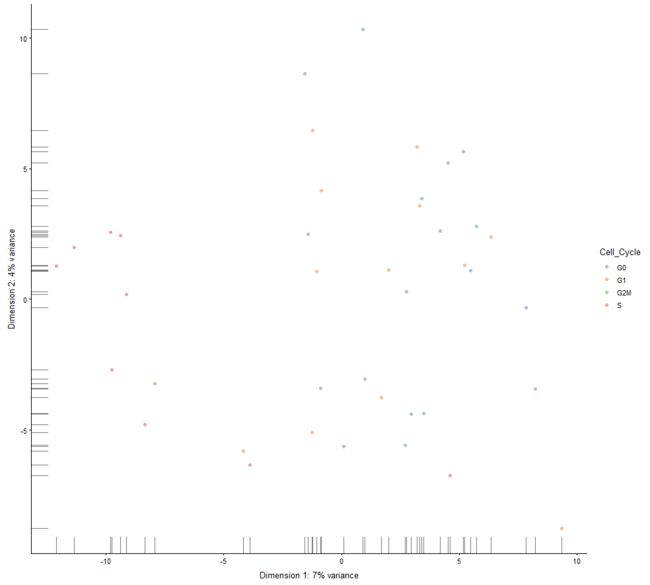
plotReducedDim(example_sce, "PCA", colour_by="Cell_Cycle")
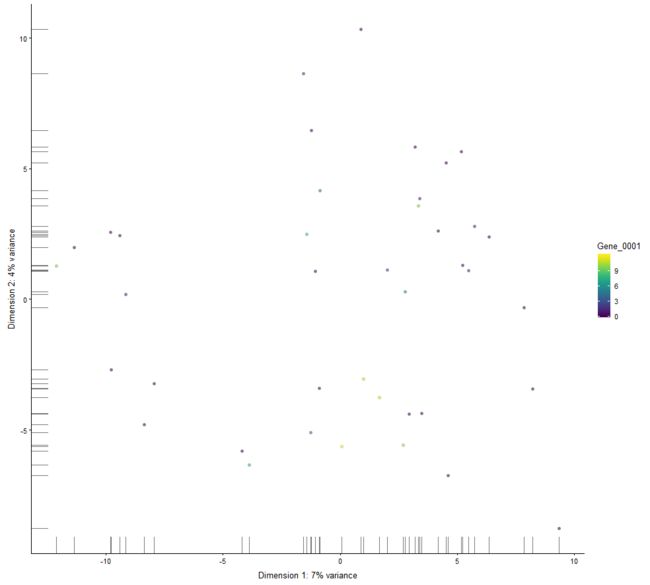
plotReducedDim(example_sce, "PCA", colour_by="Gene_0001")
plotReducedDim(example_sce, "PCA", ncomponents=5)
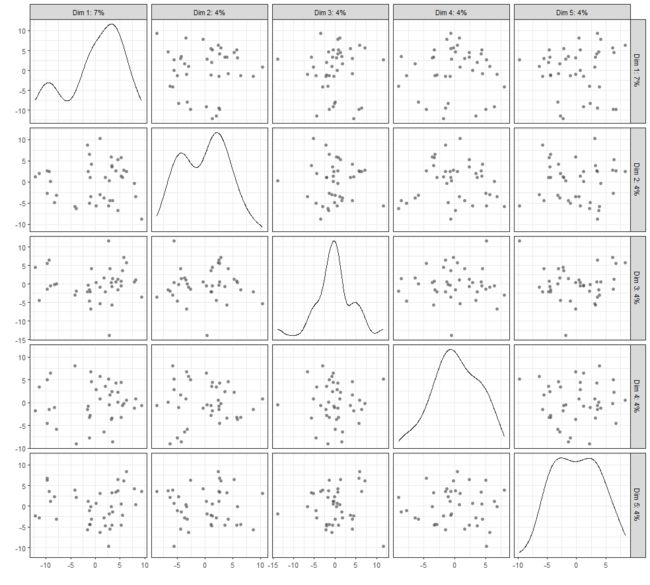
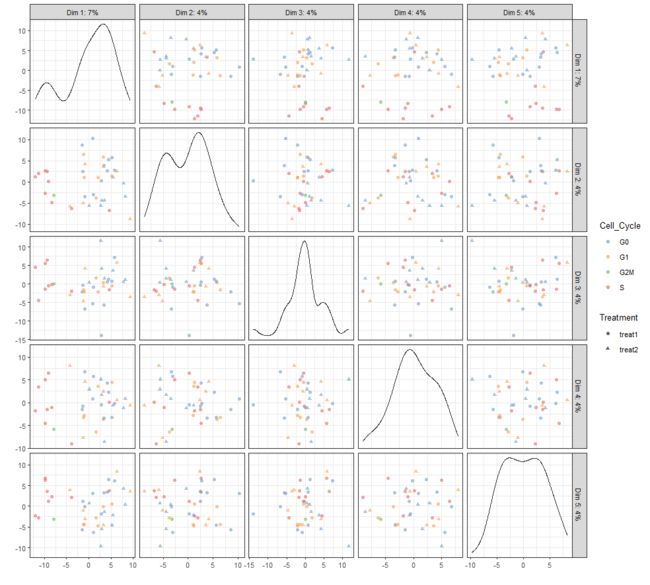
plotReducedDim(example_sce, "PCA", ncomponents=5, colour_by="Cell_Cycle",
shape_by="Treatment")
plotRLE
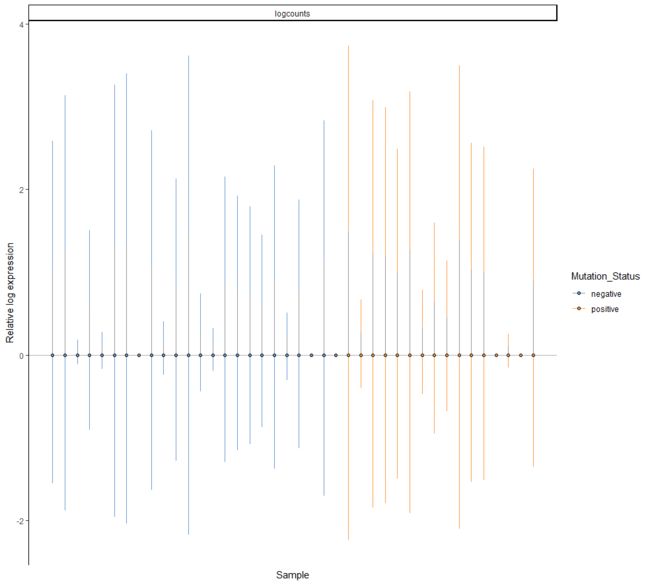
Produce a relative log expression (RLE) plot of one or more transformations of cell expression values.
data("sc_example_counts")
data("sc_example_cell_info")
example_sce <- SingleCellExperiment(
assays = list(counts = sc_example_counts),
colData = sc_example_cell_info
)
example_sce <- normalize(example_sce)
plotRLE(example_sce, colour_by = "Mutation_Status", style = "minimal")
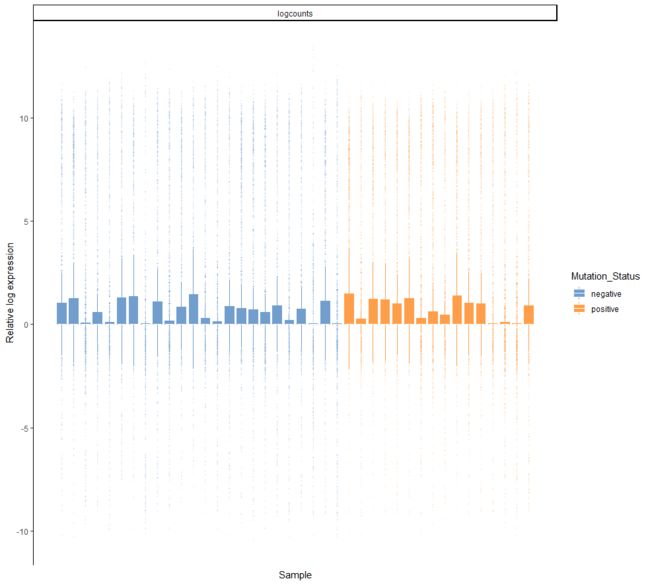
plotRLE(example_sce, colour_by = "Mutation_Status", style = "full",
outlier.alpha = 0.1, outlier.shape = 3, outlier.size = 0)
plotRowData
Plot row-level (i.e., gene) metadata from a SingleCellExperiment object.
data("sc_example_counts")
data("sc_example_cell_info")
example_sce <- SingleCellExperiment(
assays = list(counts = sc_example_counts),
colData = sc_example_cell_info
)
example_sce <- calculateQCMetrics(example_sce,
feature_controls = list(ERCC=1:40))
example_sce <- normalize(example_sce)
plotRowData(example_sce, y="n_cells_by_counts", x="log10_total_counts")
plotRowData(example_sce, y="n_cells_by_counts",
size_by ="log10_total_counts",
colour_by = "is_feature_control")
plotScater
Plot the relative proportion of the library size that is accounted for by the most highly expressed features for each cell in a SingleCellExperiment object.
## Set up an example SingleCellExperiment
data("sc_example_counts")
data("sc_example_cell_info")
example_sce <- SingleCellExperiment(
assays = list(counts = sc_example_counts),
colData = sc_example_cell_info
)
plotScater(example_sce)
[外链图片转存失败,源站可能有防盗链机制,建议将图片保存下来直接上传(img-9Y6IsBEG-1571570422795)(http://baimoc.ziptop.top/blog/20190406/smYj6xT8Fyil.png)]
plotScater(example_sce, exprs_values = "counts", colour_by = "Cell_Cycle")
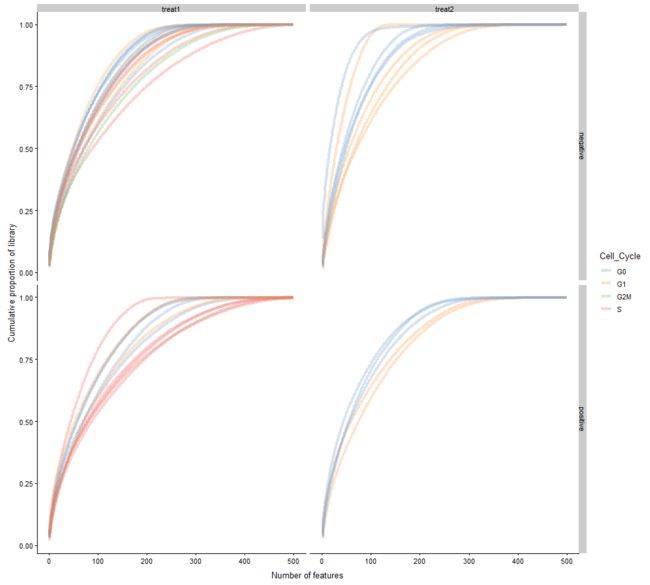
plotScater(example_sce, block1 = "Treatment", colour_by = "Cell_Cycle")
cpm(example_sce) <- calculateCPM(example_sce, use_size_factors = FALSE)
plotScater(example_sce, exprs_values = "cpm", block1 = "Treatment",
block2 = "Mutation_Status", colour_by = "Cell_Cycle")
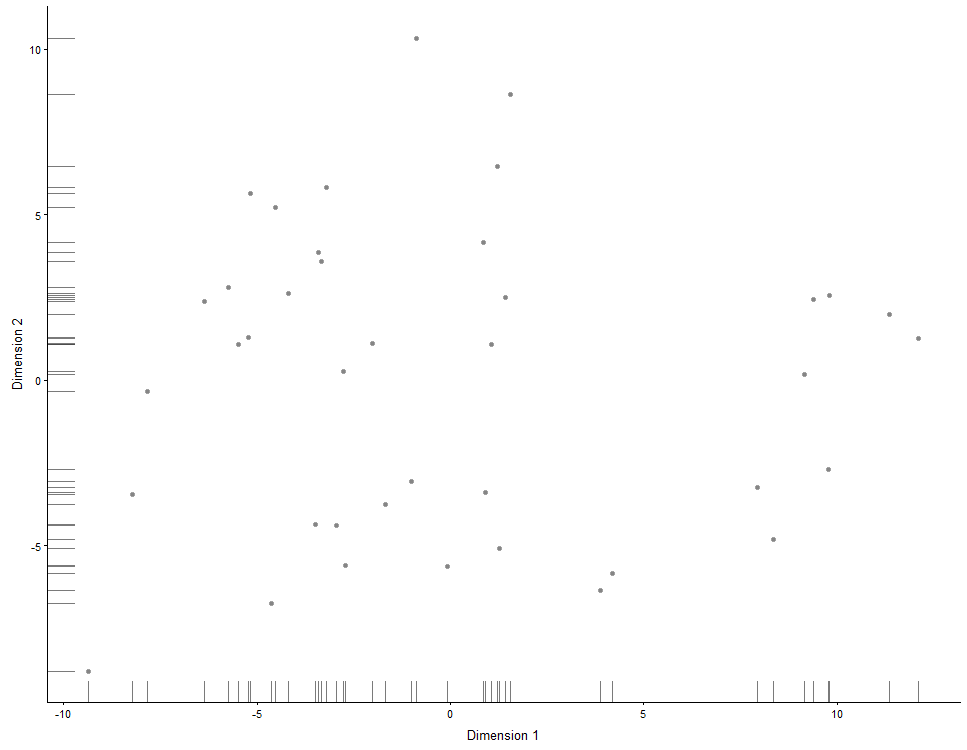
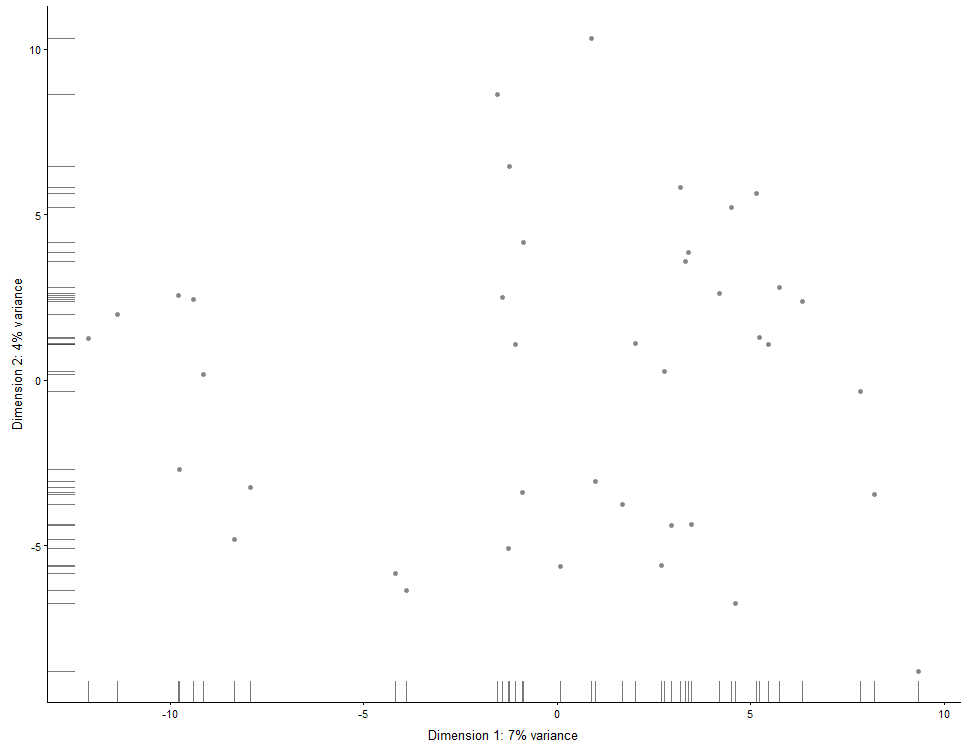
Reduced dimension plots
PCA
## Set up an example SingleCellExperiment
data("sc_example_counts")
data("sc_example_cell_info")
example_sce <- SingleCellExperiment(
assays = list(counts = sc_example_counts),
colData = sc_example_cell_info
)
example_sce <- normalize(example_sce)
## Examples plotting PC1 and PC2
plotPCA(example_sce)
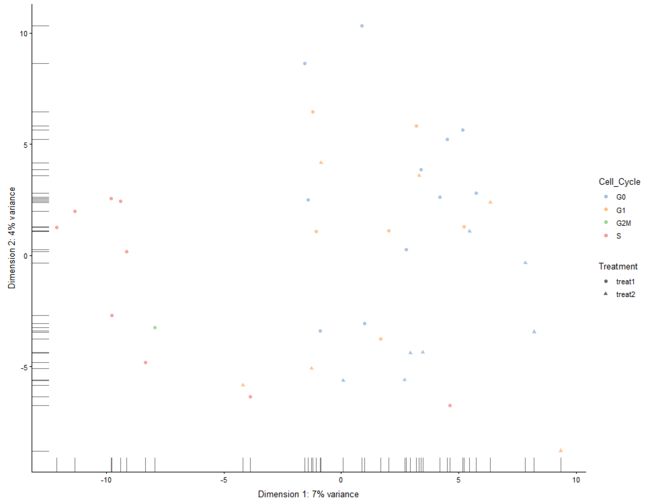
plotPCA(example_sce, colour_by = "Cell_Cycle")
plotPCA(example_sce, colour_by = "Cell_Cycle", shape_by = "Treatment")
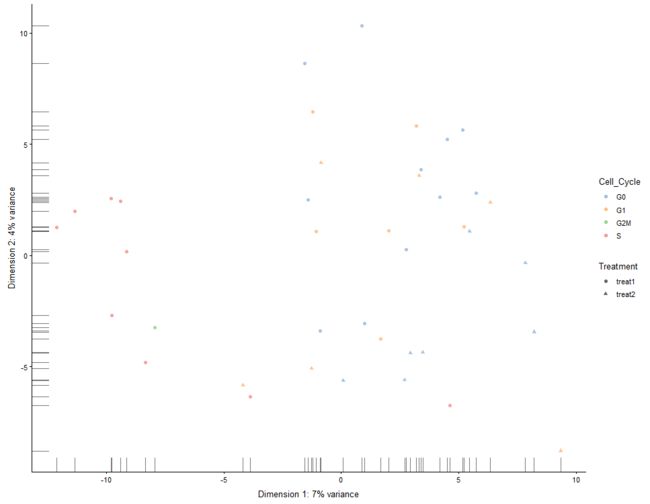
plotPCA(example_sce, colour_by = "Cell_Cycle", shape_by = "Treatment",
size_by = "Mutation_Status")
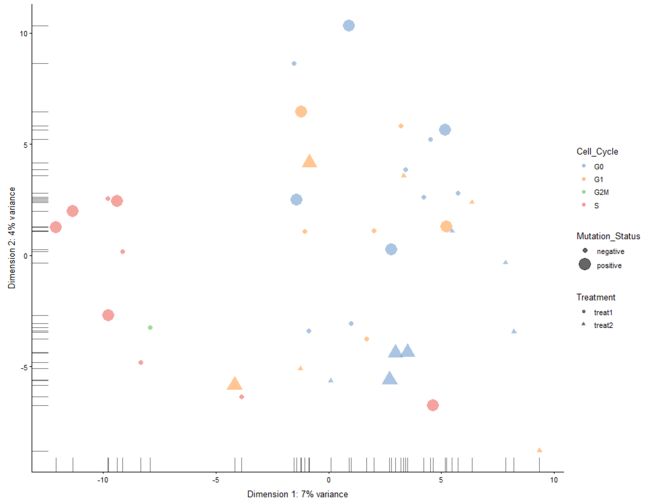
## Force legend to appear for shape:
example_subset <- example_sce[, example_sce$Treatment == "treat1"]
plotPCA(example_subset, colour_by = "Cell_Cycle", shape_by = "Treatment",
by_show_single = TRUE)
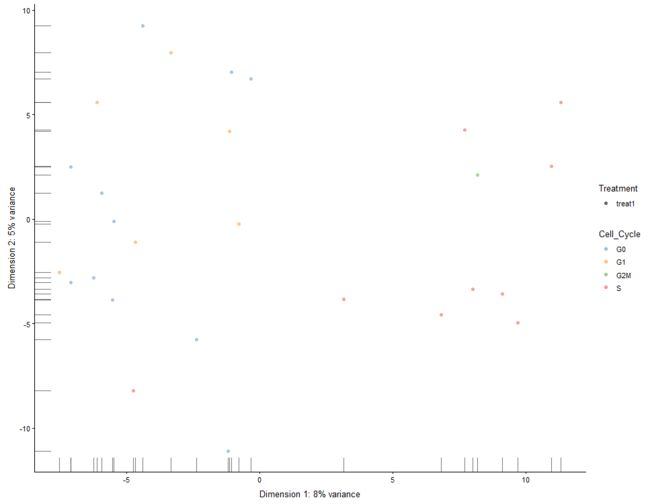
## Examples plotting more than 2 PCs
plotPCA(example_sce, ncomponents = 4, colour_by = "Treatment",
shape_by = "Mutation_Status")
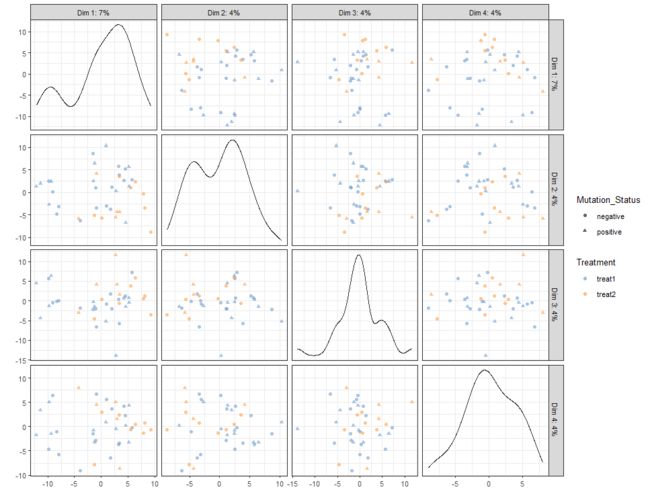
## Same for TSNE:
plotTSNE(example_sce, run_args=list(perplexity = 10))
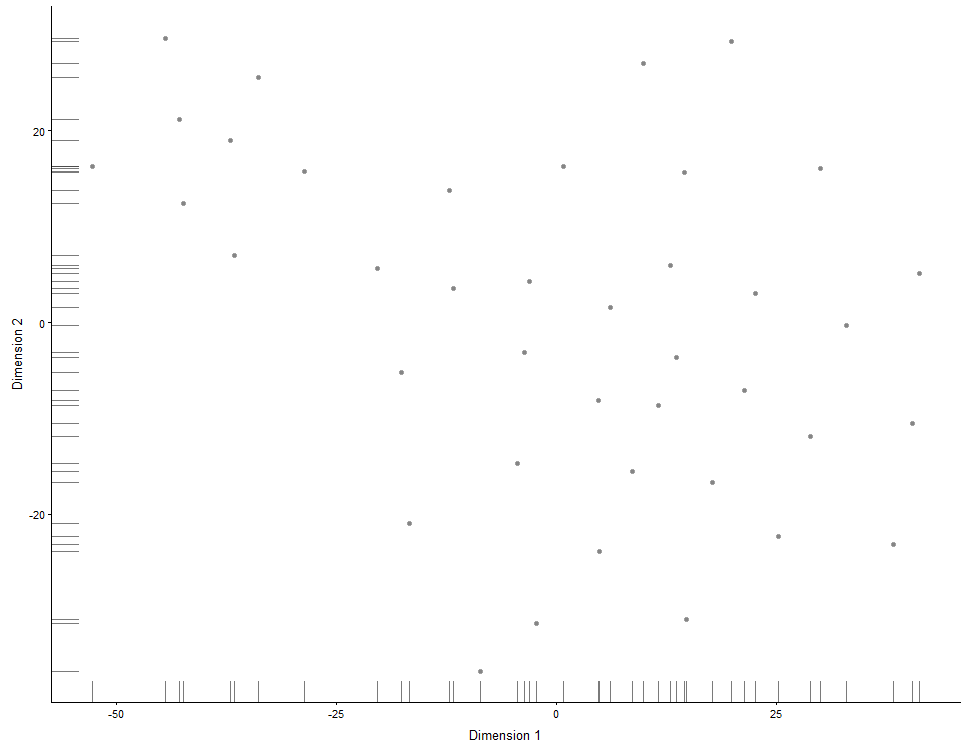
## Same for DiffusionMaps:
plotDiffusionMap(example_sce)
## Same for MDS plots:
plotMDS(example_sce)