1. What is an Over-Representation Analysis (ORA)?
ORA tries to find representative functions of a list of genes by comparing the number of times a function is observed to a baseline. Gene expression level or score were not used.
2. What are problems of the ORA analysis?
The shortcomings of the overlap analysis are that:
- ORA analysis does not account for the magnitude of expression levels. The gene is either in the list or not.
- ORA typically uses only a subset of genes - the cutoffs are "arbitrary" in the sense that they are based on convention rather than an objective measure.
- Genes and functions are all considered independent of one another. For statistical assumptions to work, this is an essential requirement. If the independence constraint does not hold then the mathematical basis for the test does not hold either. As we all know many functions in the cell are strongly interdependent.
- TAKE HOME MESSAGE: ORA analysis is more suitable for hypothesis generation than providing final answer to a problem.
Further reading: Khatri et al. "Ten years of pathway analysis: current approaches and outstanding challenges." (2012)
This review was written in 2012 so it does not contain the most up-to-date information on "pathway" analyses. But it is a good introductory material to get to learn more about the differences between different functional "pathway" analyses.
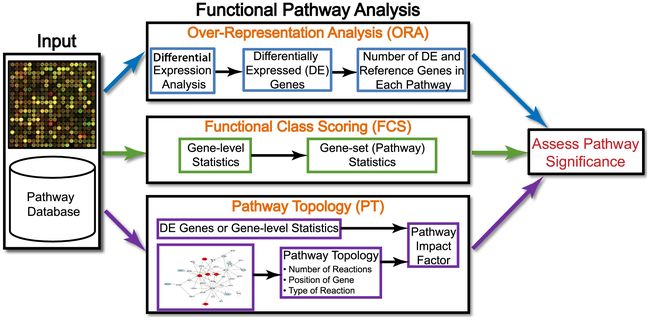
Ref: Khatri et al. "Ten years of pathway analysis: current approaches and outstanding challenges." (2012)
ermineJ (ORA, GSR, CORR) Gene set analysis tool.
ermineJ
Install ermineJ on 64 bit windows. Double-click the short-cut on desktop to start ermineJ.
Gene Set Enrichment Analysis (GSEA)
GSEA software
You will have to register to get the download link.
Turorials are also available. You can follow the tutorials to run sample data.
If you want to use your own data to run GSEA, you can follow User Guide to prepare your data. If you feel it's hard to learn, you can refer to ==Jimmy's post:=="用GSEA来做基因集富集分析" on how to run GSEA. The most import part is to prepare your data as instructed in User Guide.
clusterProfiler (ORA, GSEA analyses)
Insatllation:
## try http:// if https:// URLs are not supported
source("https://bioconductor.org/biocLite.R")
## biocLite("BiocUpgrade") ## you may need this
biocLite("clusterProfiler")
Well, this is the most well-documented software by it's owner.
Please refer to the following posts to learn how to use clusterProfiler.
1. clusterProfiler: statistical analysis and visualization of functional profiles for genes and gene clusters
2. clusterProfiler.Rmd on Github
3. 听说你有RNAseq数据却不知道怎么跑GSEA
How to prepare geneList for clusterProfiler:
If there's duplicates in your row names, you can consider using "aggregate" function to combine them and the values can be max, mean, median or min, whichever you prefer to use.
Original data: first col is gene ID (Entrez ID, but also can be other types of IDs cause you can transfer them by using bitr() function), the second column should be gene expression value or any other kind of numeric value.
d = read.csv(your_csv_file)
## assume 1st column is ID
## 2nd column is FC
## feature 1: numeric vector
geneList = d[,2]
## feature 2: named vector
names(geneList) = as.character(d[,1])
## feature 3: decreasing order
geneList = sort(geneList, decreasing = TRUE)
# Ref:https://mp.weixin.qq.com/s/aht5fQ10nH_07CYttKFH7Q
Once geneList is generated, you can use R code provided in the clusterProfiler User Manual.
Please be advised that different gene set analysis software may use different annotation files, which may greatly affect your results. Please refer to the following posts to learn more.
4. 你昨天才做的分析,可能是几年前的结果!
5. 富集分析,俩人做的结果差5岁 | 你用的注释文件有多老?
Other topics:
Recommend this review: Rhee, Seung Yon, et al. "Use and misuse of the gene ontology annotations." Nature Reviews Genetics 9.7 (2008): 509-515.
How to access Windows folders in bash Ubuntu?
C is mounted in bash Ubuntu as /mnt/c/
D is mounted in bahs Ubuntu as /mnt/d/
- How to reset you bashrc file?
Type the following in your terminal,
/bin/cp /etc/skel/.bashrc ~/
It will replace your corrupt ~/.bashrc with a fresh one. After that you need to source the ~/.bashrc so that the change take place immediately, write in terminal,
source ~/.bashrc