lachesis辅助组装流程
准备工作:
- 准备数据
- 参考基因组:Ler-1.allpaths_lg.final.assembly.fasta
- HiC数据:data_1.fastq.gz data_2.fastq.gz
- 安装所需软件并软连接到~/.local下。(添加环境变量)
- bwa
- samtools
其他支持性软件: - bedtools
- lachesis
- R
工作流程
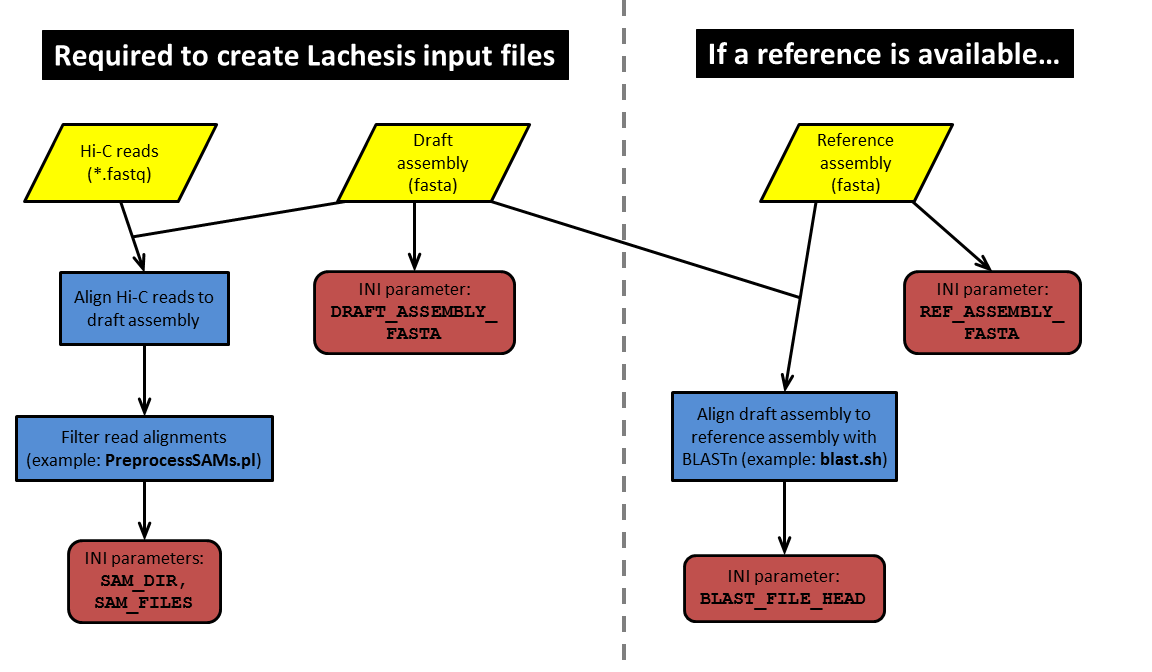
建立参考基因组的bwa索引
bwa的比对没有bowtie2那么严格。
mkdir ref && cd ref
bwa index Ler-1.allpaths_lg.final.assembly.fasta
数据比对
cd ..
mkdir 02.bwa && cd 02.bwa
bwa mem ../ref/Ler-1.allpaths_lg.final.assembly.fasta -t 10 ../01.fq/data_1.fastq.gz ../01.fq/data_2.fastq.gz > ninanjie.sam
数据过滤
- 过滤掉比对时大于2000表示分段匹配结果的sub-alignment。
perl /data/software/3dgenome/pip/LACHESIS/PreprocessSAMs-rmsubalig.pl
ninanjie.sam ninanjie.II.sam
- 过滤距离酶切位点500bp以外的reads,并选取唯一比对的reads。
这一步需要用到PreprocessSAMs.pl。我们需要用vim打开这个脚本修改一些内容以适应当前所需要处理的物种。
vim PreprocessSAMs.pl
测试物种是拟南芥,HiC实验中酶切使用的是HindⅢ,对应的酶切位点序列是AAGCTT,因此需要修改$RE_site = 'AAGCTT'(一般现在用四碱基酶比较多,因为四碱基酶的酶切位点44比六碱基酶的46更多,分辨率更高。)

这里还需要指定Lachesis内部的一个脚本
make_bed_around_RE_site.pl的位置还有
bedtools和
samtools的安装位置。另外两个
$mem和
$picard_head就注释着不用管。
接下来修改
PreprocessSAMs.sh文件
vim PreprocessSAMs.sh

这里需要将
DIR=设置成上一步
PreprocessSAMs.pl所在的文件夹,把
ASMs=设置成前面生成的
ninanjie.II.sam,
ASSEMBLY=设置成参考基因组所在的位置。
运行
PreprocessSAMs.sh脚本
sh PreprocessSAMs.sh
cd ..
到这里为止前期的数据准备阶段就完成了,接下来就是激动人心的Lachesis组装阶段了~
mkdir 03.lachesis && cd 03.lachesis
mkdir bam_file
cd bam_file
ln -s /path/to/samplex.II.REduced.paired_only.bam
ln -s /path/to/sampley.II.REduced.paired_only.bam
cd ..
复制一个test_case.ini文件到当前目录
cp /path/to/test_case.ini
这里需要根据物种的不同修改很多的参数。
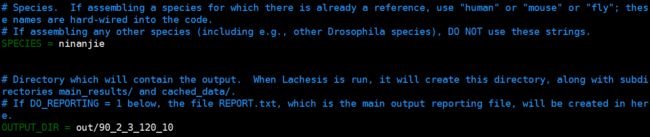
SPECIES可以不修改不影响结果。
OUTPUT_DIR设置成
out/90_2_3_120_10,这里后面的这串数字是下面要设置的各项参数,建议先把下面设置好之后再回来设置这一项。
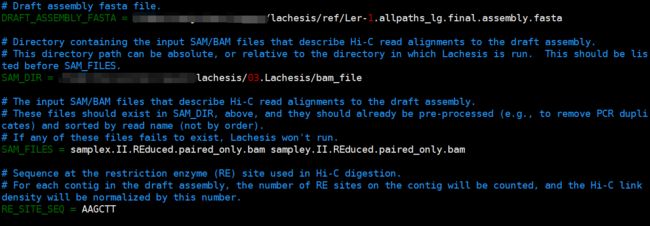
DRAFT_ASSEMBLY_FASTA = 修改为参考基因组的路径
SAM_DIR 后面改为比对文件所在的路径(即上一步的bam_file)
SAM_FILES 后面加过滤得到的bam文件的名字(拟南芥的有两组数据
samplex.II.REduced.paired_only.bam
sampley.II.REduced.paired_only.bam )
RE_SITE_SEQ 后面接酶切序列(拟南芥是
AAGCTT)
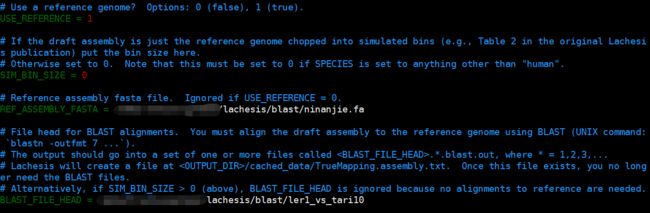
USE_REFERENCE
0代表不使用标准基因组进行评估
1表示用标准基因组进行评估
REF_ASSEMBLY_FASTA 后面是标准参考基因组(即该物种最权威的基因组版本)
BLAST_FILE_HEAD后面加标准基因组和参考基因组的blastn比对结果(我们自己的参考序列版本与权威版本TAIR10进行BLASTN比对的结果)

这里基本不用修改,如需修改请按照注释内容进行修改。

CLUSTER_N =物种所对应的染色体数目(拟南芥=5)
CLUSTER_MIN_RE_SITES(聚类分析中contig/scaffold序列中的最小酶切位点数,建议设为90)
CLUSTER_MAX_LINK_DENSITY(聚类分析中contig/scaffold序列中的最大Link深度,建议设为2)

CLUSTER_NONINFORMATIVE_RATIO(聚类回插中contig/scaffold序列与目标cluster互作数同其他cluster互作数比例,建议设为3。即待回插的序列与目标cluster的互作强度大于其他序列的3倍才予以回插)
ORDER_MIN_N_RES_IN_TRUNK( 排序分析中TRUNK内contig/scaffold序列中的最小酶切位点数,建议设为120。前面设置90的是单条contig/scaffold序列中的最小酶切位点数,这里是由contig/scaffold连接成TRUNK之后的最小酶切位点数)
以上这些参数都是需要根据不同的物种进行多次调节尝试的。
运行lachesis
mkdir ~/bin
mkdir ~/public_html
#新建的这两个目录是作为组装基因组和标准基因组比对图片的存放地址
export PATH=/path/to/R/R-3.4.1/bin/:$PATH
export PATH=/path/to/shendurelab-LACHESIS-c23474f/:$PATH
ulimit -s unlimited
#对上面这一项不熟悉的可以参考→https://zhidao.baidu.com/question/753187924640549364.html
Lachesis test_case.ini
新建的两个目录是作为组装基因组和标准基因组比对图片的存放地址。
运行需要好一会儿,建议网络不稳定的小伙伴最后一步挂nohup或者开screen。

结果展示





感觉图还是挺好看的~放到文章里也完全ok。
2018-10-1 updates:
培训的时候写的部分记录,果然有些东西就跟晚上的梦一样,一定要马上记录下来否则随着时间就渐渐消散得无影无踪了。
HiC培训问答拾遗
Q: 如何区分compartment?
A: 通过看GC含量。compartment A和compartment B会有明显的GC含量的区别。从各种图上都是无法直接得出直观的结果的,需要依据矩阵计算。
Q: loop和TAD是什么关系?
A: 他们只是不同的切入点而已。
Q: 有哪些因素会影响HiC的分辨率
A: 主要有三个:测序深度,内切酶和bin的大小。
测序深度越深,HiC分辨率越高(当然到后期提升是会逐渐趋于平缓的)。
四碱基内切酶分辨率会比六碱基内切酶分辨率更高。从概率上讲四碱基酶每44个碱基就会出现一个剪切位点,而六碱基酶则每46个碱基才会出现一个酶切位点。
bin相当于画图时候的像素点,bin的size选择得越小,数量越多,分辨率越高。
目前普遍能接受10kb左右的分辨率。
HiC的理论基础
- 染色体在核内是存在染色体疆域(Chromatin Territory)的。即染色体与染色体之间不是随意混合交缠在一起的,而是泾渭分明有自己明显的区域与界限的。实验支持:用射线破坏细胞核的某个点,受到损伤的总是部分染色体,而不是所有的染色体都受到损伤。
- 顺式作用高于反式作用。顺式作用指同一染色体内的相互作用,反式作用指染色体之间的相互作用。因为染色体疆域的存在,顺式作用会远远高于反式作用。
- 互作强度随线性距离增加而减弱。