摘要
信息基因的选择是基因表达研究中的重要问题。基因表达数据的小样本量和大量基因特性使选择过程复杂化。此外,所选择的信息基因可以作为基因共表达网络分析的重要输入。此外,尚未充分探索基因共表达网络中枢纽基因和模块相互作用的鉴定。本文提出了一种基于支持向量机算法的统计学上基因选择技术,用于从高维基因表达数据中选择信息基因。此外,已经尝试开发用于鉴定基因共表达网络中的中枢基因的统计学方法。此外,还开发了差异中枢基因分析方法,以在案例与对照研究中基于它们的基因连接性将鉴定的中枢基因分组成各种组。基于这种提出的方法,已经开发了R包,即dhga(https://cran.rproject.org/web/packages/dhga)。在三种不同的农作物微阵列数据集上评估了所提出的基因选择技术以及中枢基因识别方法的性能。基因选择技术优于大多数信息基因的现有技术。所提出的中枢基因识别方法,与现有方法相比,确定了少数中枢基因,这符合真实网络的无标度属性原则。在这项研究中,报道了一些关键基因及其拟南芥直系同源物,可用于大豆中的铝毒性应激反应工程。对各种选定关键基因的功能分析揭示了大豆中铝毒性胁迫响应的潜在分子机制。
介绍
随着快速且便宜的基因组测序技术的出现,全球许多研究机构多年来生成并存储在公共领域数据库中的巨大基因组数据[1,2]。这些数据集中的大多数与来自各种实验的基因表达有关,这些实验用于理解生物和非生物环境下物种的生物学机制的行为。对于相同的压力或相关条件,通过微阵列实验产生的数据的整合和分析对于提高所考虑的假设的灵敏度以获得有效结论是必不可少的[3]。例如,与水稻和拟南芥中不同实验有关的微阵列数据的荟萃分析揭示了高度连接的关键基因的存在,这些基因在各种生物和非生物环境下是植物防御系统的核心[4,5]。
通常,微阵列数据用于遗传网络分析中的基因选择和模块检测,其受到其基因维度较多的限制,即基因数量远远大于受试者/样品的数量[6]。因此,在适当的计算方法的帮助下,从数千个基因中选择与环境相关的大多数相关基因是很重要的。在这方面,火山绘图方法[7]在研究人员中非常受欢迎,其中通过考虑它们与类别的相关性来选择基因。然而,这种方法可能不足以发现某些特征或条件的基因之间的复杂关系[8]。此外,还有几种统计和机器学习方法,即t分数,F分数,信息增益(IG)测量,随机森林(RF)和支持向量机 - 递归特征消除(SVM-RFE)[7,9-13]也已用于基因选择。然而,在这些方法中,通过仅考虑它们与类的相关性来选择基因。在这种情况下,有可能筛选出与类别虚假相关的基因。
为了理解所选基因之间的相互关系,鉴定基因模块和特定条件的关键基因,需要进行基因共表达网络的分析。加权基因共表达网络分析(WGCNA)[14]是一种最新且流行的技术,用于破译基因之间的共表达模式。 WGCNA方法通常通过使用样本间高度相关的基因表达水平来处理基因模块的鉴定[14]。该技术已成功用于检测拟南芥,水稻,玉米和杨树中的基因组件,以应对各种生物和非生物环境[5,15-18]。此外,这种方法还导致基因共表达网络(GCN)的构建,这是一种无尺度网络,其中基因被表示为节点,edge描绘了基因之间的关联[14,19]。在这样的网络中,高度连接的基因被称为中枢基因,预计它们在生物学机制中起重要作用[20-24]。中枢基因的鉴定也有助于通过基因工程减轻植物的压力。现有方法[21-24]主要集中在中心基因鉴定上,仅基于GCN中的基因连接程度。而且,这些技术在没有任何统计标准的情况下凭经验选择这些基因。此外,文献中很少有方法可以在无标度网络中识别中心节点[22-24]。
铝(Al)毒性环境是酸性土壤作物生产的主要障碍,影响了世界上约30-40%的耕地[25]。大豆(Glycine max L.)是蛋白质,不饱和脂肪,碳水化合物和纤维的主要来源,是最重要的豆类作物之一,能够为全球人口提供营养安全。大豆优选在酸性土壤上生长,并且通过Al毒性胁迫显着降低其生产力。在酸性土壤中,铝环境导致根系生长的快速抑制,并随后抑制植物对水和养分的吸收。这增加了植物对其他环境的敏感性,导致作物生产力降低[26]。在人口爆炸和全球变暖的巨大压力下,特别是通过提高大豆的生产力来实现一般的营养安全和蛋白质安全是至关重要的。然而,一般植物中铝毒性应激反应的潜在机制,尤其是大豆,至今尚未明确解释[27]。
在该研究中,提出了一种统计技术,即Bootstrap SVM-RFE(Boot-SVM-RFE),用于选择信息基因。在该技术中,在减少基因和类别之间的虚假关联的影响之后选择基因。发现所提出的基因选择技术的性能优于现有技术,而使用三种不同的数据集进行比较。此外,还提出了用于鉴定GCN中的中枢基因的统计方法。同样,该方法在选自上述数据集的基因上进行评估,并且发现其在生物网络的无标度性质方面是优越的。此外,基于提出的中枢基因识别方法开发了R包。此外,已经尝试通过使用所提出的技术整合和分析由不同实验产生的基因表达数据集,用于鉴定大豆中的Al毒性应激反应基因。已经鉴定了负责Al毒性应激的Hub基因并且已经完成了它们的功能分析。
材料和方法
Al胁迫下的大豆微阵列实验数据集从具有平台GPL4592的Gene Expression Omnibus收集(http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GPL4592)。该平台包含使用Affymetrix Soybean Genome Array生成的37,593个探针上的3855个实验样品。在这些样品中,收集了80个与Al环境相关的样品用于进一步研究。最初,使用在R [28-30]的affy Bioconductor包中提供的Robust Multichip Average(RMA)算法处理这些收集的样本的原始CEL文件。这包括背景校正,分位数归一化总结[31]。然后选择平均≥7.1且标准偏差≤2.5的微阵列实验样品,因为在这些参数设定中观察到相关图中的颜色均匀性(S1图)。最终选择了78个样品(通过3个不同的实验产生)。有关所选样品的说明在S1文件中给出。来自RMA的log2标准化转换的表达数据用于这些选择的实验样品用于进一步的统计分析。
Bootstrap支持向量机 - 递归特征消除技术(Boot-SVM-RFE)
在这里,我们提出了一种技术,即Boot-SVM-RFE,用于从高维基因表达数据集中选择信息基因。在该方法中,基于其统计显着性,使用非参数(NP)假设检验程序来鉴定信息基因。早些时候,SVM-RFE方法被用于基因表达数据的基因排序,用于鉴定癌症负责基因[13]。在该算法中,基于SVM训练期间分类中的最小重要性,基因被单独消除。该分类问题的目标函数J定义为:其中,w是SVM计算的内核宽度。在从数据集中删除第i个基因后,使用最佳脑损伤算法[32]来近似J的变化。此外,借助泰勒级数近似[6]扩展J(直到二阶),J的值由下式给出,Δwi是由于从现有数据集中去除第i个基因而导致的权重变化和ΔJ(i)用作重量修剪标准。
这里可以注意到,成本函数J是wi的二次函数,并且两者都彼此成正比。因此,wi或J的测量提供了相同的信息。保持这一观点被用作评估第i个基因对该分类的影响的排序标准[6]。在这个过程中,基因以反向消除的方式以最小的迭代方式被消除,并且在最后准备排序的基因列表。此外,大多数基因选择方法对实验条件的小排列敏感[13]。基于高维表达数据的基因排名也可能导致假基因的选择并使选择过程不可靠[33]。因此,必须根据统计测试而不是他们的等级来选择基因。考虑到上述事实,已经提出了用于选择信息基因的检验统计量。
在该测试过程中,随机选择n个每个大小为m的自举样本,并用数据集中的可用M个样本构建SVM的训练集。然后将SVM-RFE程序应用于这些n个bootstrap样本中的每一个以获得n个基因列表及其等级。因此,每个基因将具有n个等级(每个引导一个)。让得分函数即秩得分(Rij)定义为将每个基因的这些等级转换成每个自助样本中的相应得分,其中,N表示数据集中考虑的基因总数,并且pij(1≤pij≤N)是第j个bootstrap样本中第i个基因的排名位置。在获得n个bootstrap样本的所有基因的等级评分后,需要测试以下提出的假设以选择信息基因。其中,Q是第二个四分位数。
对于第i个基因,Rj(i)(N-1≤Rj(i)≤1)是第j个自举样本(j = 1,2,...,n)的等级得分。此外,Rj(i)是随机变量(rv)。因此,Rj(i)是秩的函数,因此,其经验分布关于第二四分位数是对称的。因此,在不失一般性的情况下,我们将另一个变量rj(对于fixed i)定义为:
为了测试基因i的统计显着性(H0对H1),rj以其大小的升序排列,随后,分配等级1,2,...,n,记住它们的原始符号。设T +为正rj和T-的等级之和,为负rj的等级之和。现在,为了找到测试统计量T +的分布,另一个变量Z(k)定义为:
这里,k = {1,2,... n}。现在,变量Z(k)是独立的伯努利变量,它的阶数可以得到:
然后
基因共表达网络分析
通过使用描述基因之间关联的基因共表达测量来构建GCN [23,36]。令xi为第i个基因的表达谱,即所有微阵列样品中第i个基因的表达值。然后,计算第i和第j基因之间的基因共表达相似性度量sij作为Pearson相关系数(PCC)的绝对值[14,23],其由下式给出:第i个基因和第j个基因之间的邻接得分(aij)用sij [18]定义为:其中,β(≥1)是软阈值功率,通过使用生物的无标度属性的概念确定网络[23]。 Zhang和Horvath(2005)[14]详细讨论了确定软阈值功率的详细方法。这种软阈值方法导致加权GCN满足生物网络的无标度属性。对于Al应力和对照条件,β的值被取为8用于计算邻接得分(S2图),最佳近似无标度[36],使用R2> 0.80通过拟合幂律模型。为了鉴定所选信息基因内的基因模块(即紧密共表达基因组),基于邻接得分构建拓扑重叠矩阵[14]。执行R的WGCNA包[19]中可用的BlockWiseModules函数来识别这些模块。为此,模块尺寸,深分割水平和树合并切割高度等各种参数分别设定为20-30,4和0.15-0.25。为了找到显示跨应力和对照条件的基因的共表达模式的共有模块,功能blockwise, Consensus, Modules分别用作参数设置8,30和0.15作为功率,最小模块尺寸和合并切割高度。
提出用于鉴定中枢基因的统计方法
在网络理论中,如果节点的连接度大于网络的平均连接度,则节点被定义为中心节点[20-24] [21]。在现有方法中,基因被指定为基于指示功能的中枢基因[21,23],即Hubi = [I(ki>τ)],并且遗传网络中枢基因(NHub)的数量计算为,其中,Hubi:第i个基因的中心状态(即1或0); ki:第i个基因的连接程度; τ:阈值即网络的平均连接度。该技术仅基于观察到的基因连接性凭经验选择中枢基因,而不考虑任何统计学考虑。因此,提出了一种基于基因连接统计显着性的替代统计方法,用于检测GCN中的中枢基因。拟议的统计方法如下:
根据加权基因连接(aij),第i个基因的加权基因评分(WGS)可写为:其中,WGSi表示基于其与GCN中剩余基因的连接的第i个基因的相对重要性。为了进行中枢基因鉴定,构建了以下假设。其中,μ是完整网络模型的平均连接度。这里为了在H0下获得测试统计量的分布,使用了重采样过程。在该过程中,从M个微阵列样品中以相等的概率随机选择m个微阵列样品以构建一个子样品(对于一个GCN)(m≤M)。然后应用统计测量(方程13-15)来获得该GCN中每个基因的WGS。这个程序重复多次说S得到S组WGS。在该研究中,S = 500用于分别在应激和控制条件下获得500个随机GCN。为了测试H0对H1,提出了NP检验统计量来测试每个基因的WGS的显着性,即用于测试基因的WGS是否大于完整网络的平均连接程度。建议的假设检验程序如下:
对于特定基因(i),WGSk(i)是第k个子样本(k = 1,2,...,S)的WGS。这里WGSk(i)是rvs。因此,在不失一般性的情况下,另一个变量Xk可以定义为:
为了测试基因i的连通性的统计显着性,Xk以其大小的升序排列,随后,分配等级1,2,...,S,记住Xk的原始符号。设W +为正Xks和W-的等级之和,为负Xks等级的总和。 H0下的检验统计量(W +)的分布可以通过上述方法得到,以获得Boot-SVM-RFE中T +的分布。此外,在大量子样本(S = 500)下,遵循中心极限定理,W +的分布近似为标准正态分布,即
对GCN中的每个基因重复该过程,并且应用统计检验以分别基于对照和胁迫条件的显着性值鉴定中枢基因。
算法:
步骤1:从GCN中的所有基因(节点)开始
步骤2:构建数据集,称为Tk,其中m个样本随机取自M个微阵列样本
步骤3:计算所有基因的WGS
步骤4:重复步骤2和3次S,得到每个基因的S组WGS
步骤5:获取特定基因(第i个基因)及其WGS
步骤6:测试第i个基因的假设并获得其p值
步骤7:对所有基因重复步骤5-6(i = 1,2,...,G)
步骤8:对p值进行排序并选择中枢基因
在两种对比条件(实验与对照)下构建的GCN的所提出的中枢基因识别方法可称为差异中枢基因分析(DHGA)。通过这种方法,基于显着性的统计检验,在这两种GCN中可以鉴定中枢基因。在p值的基础上,在任一条件下GCN中的基因可以分组成不同的组,即。管家中心基因(HHG,用于压力的独特中枢基因(UHG,用于控制的UHG,基于决策矩阵的非中枢基因(表2)。
模块交互网络建模
模块交互网络(MIN)可以定义为有向图,其中模块是节点或单元,边缘描述模块之间的调节关系。由于高维度问题,文献中可用于遗传网络建模的大多数方法不适用于基因表达数据[37]。将基因模块视为功能单元在生物学上是必需的[18,20]。此外,这些模块可以作为研究基因模块之间相互作用的单元。模块的表达水平可以计算为:其中,Md(t):时间t(t = 1,2,...,T)的第d个模块(d = 1,2,...,D)的表达水平,Gi(t):时间t的第i个基因的表达水平,nd:第d个模块中存在的基因数。
对于Al胁迫数据,通过荟萃分析获得的所有选择的微阵列样品属于时间序列实验(基因表达值在5个时间点测量,例如0,2,12,48和72小时)(S1文件)。此外,数据插值的bspline方法用于在[0,72小时]的间隔内插入最多50个时间点的模块表达值。然后,我们在时间t将模块d的表达水平建模为在时间(t-1)处具有其他模块的表达水平的线性回归。描述这些模块之间相互作用的模型可以写成:其中,βh是回归系数,ε是随机噪声,均值为0,方差为σ2。为了计算描述模块之间相互作用的回归系数,执行贝叶斯模型平均(BMA)[38]算法。此外,通过使用迭代BMA算法[39]计算每个模块交互的后验概率。模块相互作用按后验概率值的降序排列,通过固定适当的阈值选择显着的模块相互作用。 MIN是使用RCytoscape包[40]构建的。
结果
Boot-SVM-RFE的性能分析
为了研究所提出的Boot-SVM-RFE技术的性能,基于来自每种基因选择技术的分级获得的前1000个基因用于通过SVM分类器将作物微阵列样品分类为对照和胁迫类别。测量每个滑动窗口尺寸超过5倍交叉验证的CA.表3中给出了不同滑动窗口尺寸的CA.观察到对于Al应力数据,Boot-SVM-RFE的CA高于其他技术的CA.滑动窗口尺寸为50,100,150,200,250和300的SVM-RFE,t-score,F-score,RF和IG。但是,对于更高的窗口尺寸,即350,400,450和500,CA为Boot-SVM-RFE与SVM-RFE相当,但高于t-score,F-score,RF和IG(表3)。通常,Boot-SVM-RFE的性能最高,其次是相对于Al应力的CA值的SVM-RFE,RF,IG,F-得分和t-得分。然而,在盐度和冷应力的情况下,无论滑动窗口的大小如何,Boot-SVM-RFE的性能都优于其他基因选择技术(表3)。在盐度胁迫的情况下,不同基因选择技术的表现顺序是Boot-SVM-RFE> SVM-RFE> F得分> RF> F得分> t得分> IG,而在冷应激的情况下,表现顺序是Boot-SVM-RFE> RF> IG> SVM-RFE> F得分> t得分(表3)。从该性能分析可以看出,所提出的Boot-SVM-RFE的性能在与非生物胁迫相关的不同数据集中始终优于其他现代技术。此外,还观察到,当应用于这三种应力的数据集时,Boot-SVM-RFE相对于其他五种技术对于大多数滑动窗口尺寸具有较小的CV(表3)。
大豆铝环境条件下信息基因的筛选
由于发现Boot-SVM-RFE与其他基因选择技术相比更优越,因此进一步用于选择大豆中Al胁迫的信息基因。 为了获得稳健且最小的信息基因组,将Volcano图中的倍数变化替换为从Boot-SVM-RFE获得的-log10(p值),然后构建基因选择图。 基因选择图的Y轴和X轴的阈值分别固定为4和2.5,这导致981个基因的选择(图1)。 然后使用从GeneChip Soybean Genome Array of Affymetrix获得的这些981基因的共有序列来鉴定拟南芥直系同源物[41],并且发现554个基因在拟南芥中具有独特的直系同源物(图1)。 此外,这些选定基因的注释来自SoyBase(http://soybase.org)[42]。 S1表中给出了关于这些所选基因的简要描述。
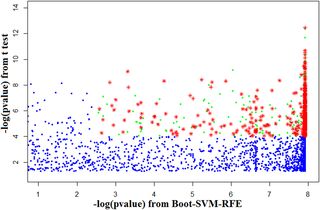
图1.选择大豆铝胁迫信息基因的基因选择图。
横轴表示从Boot-SVM-RFE获得的统计显着性值的负对数。 纵轴显示来自t检验的统计显着性值的负对数。 绿点表示选择的探针,其中-log(p值)来自Boot-SVM-RFE≥2.5的阈值和t-检验 - 对数(p-值)≥阈值4.红色星形表示所选择的具有拟南芥直向同源物的探针。 蓝点表示未选择的探针。
大豆铝胁迫选择基因的功能分析
通过使用AgriGO [43],植物特异性GO术语富集分析工具,对981个选定基因进行基因本体论(GO)富集分析。观察到大多数选定的基因负责过渡金属离子结合,金属离子结合,阳离子结合,离子结合等(图2A)。由于水或土壤中高浓度的Al离子,这些分子功能(MF)可能被激活。另外两种MF即氧化还原酶(氧化还原酶)和激酶活性也存在于这些选定的基因中(图2A)。基因在氧化还原活性中的显着行为可能与复杂化学反应中的电子传递有关,其在离子传输期间平衡电荷。氧化还原活性也可能与活性氧物种(ROS)有关,这些物质是由于在非生物胁迫(如铝毒性应激)中由于缺水导致的氧化应激而产生的[44]。在生物过程类别,如细胞氮化合物代谢过程,胺代谢过程,细胞氨基酸和衍生物代谢过程,含氧酸代谢过程,有机酸代谢过程,羧酸代谢过程,细胞酮代谢过程和离子转运活性,数量选择的基因与其他生物过程相比更多(图2A)。可以推断,这些选择的基因中的一些参与离子转运活性,即参与将离子转运到细胞外以维持细胞中的适当pH [45]。在细胞成分的情况下,选择的基因与转录因子复合物,细胞质膜结合的囊泡,膜结合的囊泡,细胞质囊泡,囊泡和核质部分相关(图2A)。可以看出,基因的最大数量与囊泡和膜有关,这与Al胁迫条件下可获得的金属离子的解毒机制一致,特别是在液泡的隔离中[46,47]。发现膜上存在的一些选定基因参与金属离子在细胞外或液泡中的转运,以维持pH和跨膜质子梯度[48]。

图2. Al胁迫下所选基因和hub基因的功能富集分析。
对于不同的基因本体类别(CC,MF和BP),显示了使用Agrigo对于Al胁迫条件的981个选择的信息基因(A)和中枢基因(B)的GO术语富集分析。 对于(A),选择其GO值<0.008和FDR值(错误发现率)<0.6的GO项。 对于(B),选择其GO值<0.1且FDR值<0.8的GO项。
大豆铝环境下的基因共表达网络分析
使用WGCNA,将选定的981个基因分别分为19个和18个模块(包括灰色模块,它们是非模块基因的模块),分别用于Al胁迫和控制条件(图3)。在这两种情况下,由绿松石颜色表示的模块包含最大数量的基因,因此被指定为任一条件的最大模块。基于Al胁迫和对照条件的这些选定基因的表达谱,获得23个共有模块(具有相似共表达模式的基因组)。任何条件的各种模块与共识模块的匹配可以从图3中以其颜色可视化。在S3中给出了共识与对照和共识与应力条件的各个模块之间的串扰程度。此外,树状图中的长分支和热图中的高强度红色(S4图)显示与存在于模块外部的基因相比,属于同一模块的基因具有更高程度的共表达。表4给出了每个模块的模块成员资格(存在的基因数)及其在Al胁迫条件下的基础分子功能。观察到,每个模块都用GO术语进行了显着注释,除了以绿黄色表示的基因模块和灰色(表4)。因此,可以推断出这两个模块中存在的基因的功能仍然很大程度上是未知的。此外,19个识别的模块(包括灰色模块)被用作模拟MIN的功能单元。计算每个模块相互作用的后验概率(使用iBMA算法)并用于构建Al应力的MIN,如图4所示。可以看出模块1和18与MIN中的大多数模块相互作用(图4) )。因此,这些模块及其成员可能在大豆的Al胁迫响应中发挥重要作用。

图3. Al环境和对照条件下所选基因和基因组件的聚类树状图。
表示了共识模块(CM)与应力(SM)(A)和控制(NM)(B)条件下的模块之间的对应关系。
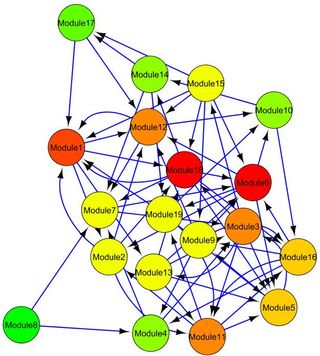
图4. Al胁迫下基因模块的模块交互网络。
该网络由19个节点和70个边缘(监管关系)组成。 为了消除模块之间的弱相互作用,后验概率的阈值固定为0.2。
提出的中枢基因检测方法的性能分析
基于现有方法(即单独的WGS),GCN中的39.05%和36.91%基因分别被检测为大豆中的应答和对照条件的中枢基因(表5)。因此,基于现有方法,大比例的基因被鉴定为GCN中的中枢基因,这与生物网络的无标度特性相矛盾(因为GCN是无标度网络)[20,36]。对于水稻中的盐度和冷胁迫,观察到类似的发现(表5)。然而,在提出的方法(计算p值)的情况下,GCN中只有23.24%和19.14%的基因被发现分别是大豆中枢基因(p值<1E-10)的压力和控制情况(表5) )。此外,通过降低所提出的方法中的重要性水平,可以进一步减少集线器的数量。这表明所提出的方法将能够识别相对较小的基因子集作为GCN中的枢纽,即较少的WGS在统计学上是显着的。对于水稻中的盐度和冷胁迫获得了类似的结果(表5)。
WGS的分布(即重右尾分布)包含较低和较高的值,其在基因的低和高连接程度之间没有太大的区别(图5)。相反,从p值的分布来看,具有高连接度的基因与GCN中低连接度的基因很好地分离(图6)。换句话说,从图6中可以更好地看出GCN中统计上强弱连接的基因之间的区别,而不是图5.此外,从提出的DHGA方法获得的两个对比条件(压力与对照)的结果给出了表6.DHGA方法允许将GCN中的水稻187(胁迫)和208(对照)中枢基因分组为HHG(141),UHG至盐胁迫(46)和UHG至对照(67)(表6)。对大豆中的铝胁迫和水稻中的冷胁迫可以进行类似的解释。
讨论
所提出的Boot-SVM-RFE技术被发现优于从高维基因表达数据中选择信息基因。这种方法也优于传统的基因选择技术,如t检验和F-score,因为它不需要任何关于数据的分布假设。在该技术中,将p值分配给每个基因,并且将具有较低p值的基因视为对所研究的特定条件/性状提供信息。与其他技术相比,基于p值的信息基因的选择对于实验生物学家来说具有科学性和统计学意义。此外,该技术中使用的自举程序有望消除基因类别的虚假关联。对比分析表明,Boot-SVM-RFE在现有技术上表现优于现有技术,即SVM-RFE,t-score,F-score,RF和IG就CA而言。此外,由于CA中所有窗口大小的CV值较低,因此其性能可被视为稳健。
基于对基因连接的统计学显着性的评估,所提出的用于中枢基因鉴定的统计方法允许GCN中候选中枢基因的排序和选择。这是通过基于随机重采样的程序完成的,其中基于NP测试计算统计显着性值,其不需要数据的高斯假设。此外,具有较低p值的基因代表GCN中高度连接的基因,因此被指定为中枢基因。此外,该方法中使用的随机化程序允许人们测试观察到的基因连接是否偶然地大于预期的基因连接值(即,拒绝随机关联的零假设)。这也能够消除基因之间的虚假关联,因为这些关联是在PCC的基础上测量的。基于p值而不是单独使用WGS选择中枢基因似乎更具统计学意义,因为与WGS相比,p值基于统计学标准提供了基因连接的可靠测量(较低的p值表示高基因连接,反之亦然)。此外,检测到的中枢基因倾向于具有更高的连接度并且与GCN中具有低连接度的基因广泛分离。此外,基于该方法,与现有方法相比,GCN中的一些以及重要基因被鉴定为中枢,其与现有方法一致,这符合生物网络的无标度特性。
使用DHGA方法,基于这两种对比条件的计算p值,将GCN中的基因分组为各种类别,如HHG,非中枢,UHG用于应激和控制。这些鉴定的中枢基因可被视为进一步研究的生物标志物,包括分析它们参与多种细胞机制。此外,HHG可用于维持对细胞存在至关重要的基础细胞功能[49],而UHG可用于作物中的应激反应工程,用于开发耐胁迫品种。
了解大豆中的铝环境响应机制对于植物育种者开发抗铝品种至关重要。在公共领域数据库中,很少有与大豆中铝环境相关的样本,这些样本是通过多项研究在不同的实验条件下产生的。因此,进行荟萃分析以组合这些数据集,并且元数据用于进一步的统计分析。然后,应用开发的技术来识别负责任的基因,以了解该作物中的胁迫响应机制。据报道,植物中Al胁迫响应涉及两个主要过程(i)从根细胞中排除Al离子和(ii)植物细胞中Al离子的解毒[50]。发现一些选定的基因参与细胞外金属离子的运输,这可能与第一个过程有关。在金属离子传输期间平衡电荷的化学反应下与电子传递有关的氧化还原活性[45]的功能可能与第二个过程有关。氧化还原活性也可能与植物响应Al胁迫产生的ROS产生有关。此外,ROS还通过脂质过氧化[51],蛋白质和核酸[52]严重破坏细胞的正常代谢。氧化还原活性的增加与抗氧化酶如过氧化氢酶,抗坏血酸过氧化物酶和愈创木酚过氧化物酶在非生物胁迫条件下的活化一致[53]。已知BP分类学中的活性如细胞葡聚糖和细胞氨基酸代谢过程在植物中响应各种非生物胁迫而增加[54],并且需要在Al毒性胁迫的背景下研究其他报道的生物过程。