网络爬虫
(web crawler),也叫网络蜘蛛(spider),是一种用来自动浏览万维网的网络机器人。其目的一般为编纂网络索引。各大搜索引擎都可以被看做爬虫,根据爬取的内容更新自身的网站内容或其对其他网站的索引。一般如果想批量从网页获取数据,有download或者API(之前推送过使用API提取TCGA数据)页面最好,没有的话可以考虑使用爬虫爬取。
本期使用R语言批量爬取NCBI基因注释信息,主要用到了XML包的getNodeSet函数。需要使用者有一定html+css基础,以及理解并能使用XML路径语言(xpath)。
使用R爬取NCBI人类基因信息流程如下:
首先准备目标基因文件,我们以下面这几个基因(gene symbol的形式)为例进行爬取其在NCBI(gene)中的信息,基因列表文件可以从这里下载(https://pan.baidu.com/s/1c2jbvby)。
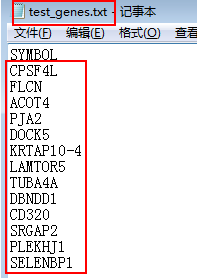
载入要用到的包并读入基因列表:
library(RCurl)
library(stringr)
library(XML)
library(clusterProfiler)
rm(list=ls())
# 读入基因列表:
genes <- read.table("test_genes.txt",header = T,stringsAsFactors = F)
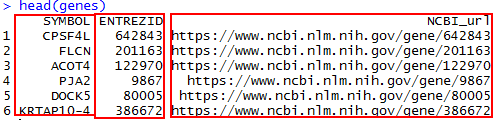
从下图可以发现NCBI对于基因页面的索引方式都是 https://www.ncbi.nlm.nih.gov/gene/Entrze ID 的方式。

所以我们需要将gene symbos转为entrze ID,这里使用clusterProfiler包的bitr函数进行转换:
# 将gene symbol转为entrze ID:
genes <- bitr(genes$SYMBOL, fromType="SYMBOL", toType="ENTREZID", OrgDb="org.Hs.eg.db")
然后获得每个基因在NCBI中的索引链接:
# 网址数据框:
genes$NCBI_url <- paste("https://www.ncbi.nlm.nih.gov/gene/",genes$ENTREZID,sep="")
head(genes)
使用XML包的getNodeSet()函数需要两个参数,一个是根据URL获得的网页XML document对象,另一个是要定位的节点(xpath格式)。不了解xpath的可以点击页面左下角阅读全文查看其基本语法。不过我们可以在不了解语法的情况下获得要定位节点的xpath。只需要在chrome浏览器里打开NCBI的gene信息页面,我们以基因DBNDD1为例,然后再按一下F12就可以调出chrome浏览器自带的开发者工具:

比如说我们要爬取基因的Official Full name信息,我们只需要在调出来的开发者工具栏右上角点几下那个小箭头,然后在点下Official Full name然后在右上方的源代码显示Official Full name的位置点击右键,选择Copy,Copy XPath。

我们可以得到这样的xpath字段:
# Official Full name的xpath://*[@id="summaryDl"]/dd[2]/text()
使用同样的方法,我们可以获得基因的HGNC ID,Gene type和Summary等任何部分的xpath。
# HGNC ID的xpath://*[@id="summaryDl"]/dd[3]/a
# Gene type的xpath://*[@id="summaryDl"]/dd[5]/text()
# Summary的xpath://*[@id="summaryDl"]/dd[10]/text()
到这里准备工作就结束了,接下来构建并调用函数来爬取每个基因这4个字段的信息:
# 根据xpath获取节点内容:
getNodesTxt <- function(html_txt1,xpath_p){
els1 = getNodeSet(html_txt1, xpath_p)
# 获得Node的内容,并且去除空字符:
els1_txt <- sapply(els1,xmlValue)[!(sapply(els1,xmlValue)=="")]
# 去除\n:
str_replace_all(els1_txt,"(\\n )+","")
}
# 处理节点格式,为character且长度为0的赋值为NA:
dealNodeTxt <- function(NodeTxt){
ifelse(is.character(NodeTxt)==T && length(NodeTxt)!=0 , NodeTxt , NA)
}
使用一个for循环获得每个基因的信息并存储到数据框:
for(i in 1:nrow(genes)){
# 获得网址:
doc <- getURL(genes[i,"NCBI_url"])
cat("成功获得网页!\t")
# 获得网页内容
html_txt1 = htmlParse(doc, asText = TRUE)
# 获得Full Name:
genes[i,"FullName"] <- dealNodeTxt(getNodesTxt(html_txt1,'//*[@id="summaryDl"]/dd[2]/text()'))
cat("写入基因\t")
# 获得HGNC ID:
genes[i,"HGNC_ID"] <- str_replace_all(dealNodeTxt(getNodesTxt(html_txt1,'//*[@id="summaryDl"]/dd[3]/a')),"HGNC|:","")
cat("写入HGNC_ID\t")
# 获得Gene type:
genes[i,"GeneType"] <- dealNodeTxt(getNodesTxt(html_txt1,'//*[@id="summaryDl"]/dd[5]/text()'))
cat("写入GeneType\t")
# 获得summary:
genes[i,"Summary"] <- ifelse(length(getNodesTxt(html_txt1,'//*[@id="summaryDl"]/dd[10]/text()'))!=0,getNodesTxt(html_txt1,'//*[@id="summaryDl"]/dd[10]/text()'),NA)
cat("写入Summary\n")
print(paste("完成第",i,"个了!"))
}
爬取结果如下:
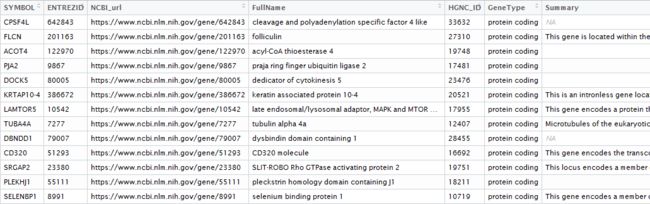
上面的节点的xpath中的标签是按照顺序在chrome生成的,这样就存在一个问题,如果某个基因没有某个属性,则这个属性后续的所有节点的xpath都将发生改变。如下图所示的两个基因,一个有别名,一个没有别名,则这两个基因的Summary的xpath就是不同的,而我们是按照有别名基因的xpath爬取的,所以爬取到没有别名的基因时的summary就会出错。

为了能够精确爬取到想要的数据,这里就需要使用到xpath的轴和函数获得准确的节点定位。下面直接附上代码:
# xpath精确定位:
for(i in 1:nrow(genes)){
# 获得网址:
doc <- getURL(genes[i,"NCBI_url"])
cat("成功获得网页!\t")
# 获得网页内容
html_txt1 = htmlParse(doc, asText = TRUE)
# 获得Full Name:
genes[i,"FullName"] <- str_split(dealNodeTxt(getNodesTxt(html_txt1,'//*[@id="summaryDl"]/dd[preceding-sibling::dt[contains(text(),"Symbol") and position()=1 ] ]')),"provided")[[1]][1]
cat("写入基因\t")
# 获得HGNC ID:
genes[i,"HGNC_ID"] <- str_replace_all(getNodesTxt(html_txt1,'//*[@id="summaryDl"]/dd[preceding-sibling::dt[text()="Primary source" and position()=1 ] ]')," |HGNC|:","")
cat("写入HGNC_ID\t")
# 获得Gene type:
genes[i,"GeneType"] <- dealNodeTxt(getNodesTxt(html_txt1,'//*[@id="summaryDl"]/dd[preceding-sibling::dt[text()="Gene type" and position()=1 ] ]'))
cat("写入GeneType\t")
# 获得summary:
genes[i,"Summary"] <- dealNodeTxt(getNodesTxt(html_txt1,'//*[@id="summaryDl"]/dd[preceding-sibling::dt[text()="Summary" and position()=1 ] ]'))
cat("写入Summary\n")
print(paste("完成第",i,"个了!"))
}
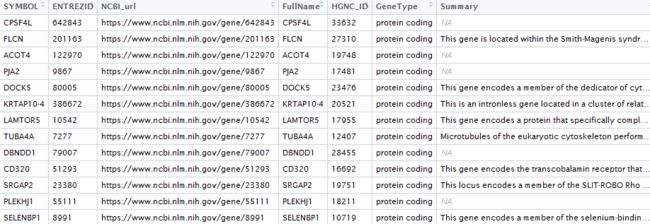
精确爬取结果如下,验证都是正确的。
爬取结果的准确性依赖于节点定位是否准确,定位既可以通过xpath,也可以通过CSS,rvest包提供里这两种定位方式。并且rvest包使用magrittr包的%*%操作符,增强了代码的可读性。
更多原创精彩视频敬请关注生信杂谈:

阅读原文