差异分析流程(二)limma
- 1. 制作三个矩阵
- 2. 建模前归一化
- 3. 建模及结果
- 4.检查
参考链接:
https://www.bioconductor.org/packages/devel/workflows/vignettes/RNAseq123/inst/doc/limmaWorkflow_CHN.html
#加载包和数据
if (!requireNamespace("BiocManager", quietly = TRUE))
install.packages("BiocManager")
if (!require("edgeR", quietly = TRUE))
BiocManager::install("edgeR")
options(stringsAsFactors = F)
load("expr.RData")
expr.RData储存了经过处理后的表达矩阵和分组信息,详见差异分析流程(一)数据预处理
1. 制作三个矩阵
# 表达矩阵
data = expr
# 分组矩阵
group_list = factor(group_list)
design <- model.matrix(~0+group_list)
rownames(design) = colnames(data)
colnames(design) <- levels(group_list)
# 差异比较矩阵
cont.matrix <- makeContrasts(contrasts = c('trt-untrt'), levels = design)
2. 建模前归一化
limma输入数据一定要log2转化原因:
https://blog.csdn.net/tuanzide5233/article/details/88542805
- 方法1: 通过boxplot查看数据整齐与否再决定是否归一化
library(RColorBrewer)
dge <- DGEList(counts = data)
col <- brewer.pal(ncol(dge$counts), "Paired")
par(mfrow=c(2,2))
boxplot(dge$counts,outline=F, col=col)
title(main="A. Unnormalised ",ylab="raw count")
boxplot(calcNormFactors(dge, method = "TMM")$counts,outline=F,col=col)
title(main="B. TMM ",ylab="raw count")
boxplot(cpm(dge$counts),outline=F, col=col)
title(main="C. CPM ",ylab="cpm")
boxplot(cpm(dge$counts,log=TRUE),outline=F, col=col)
title(main="D. Log-CPM ",ylab="log-cpm")
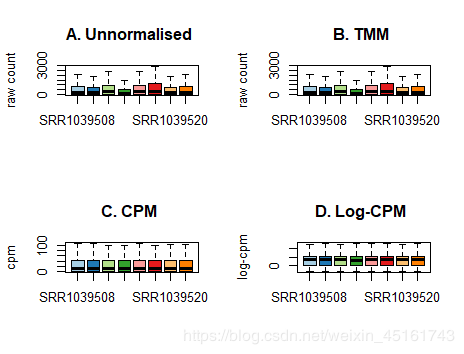
由上图可以发现log-cpm归一化效果最好,如果直接log-cpm化效果还不是很好,可以先通过TMM标准化再进行log-cpm化
- 方法2: 利用voom进行归一化
dge <- DGEList(counts = data)
dge <- calcNormFactors(dge)
v <- voom(dge, design, plot=TRUE) #会自动计算log(cpm)值
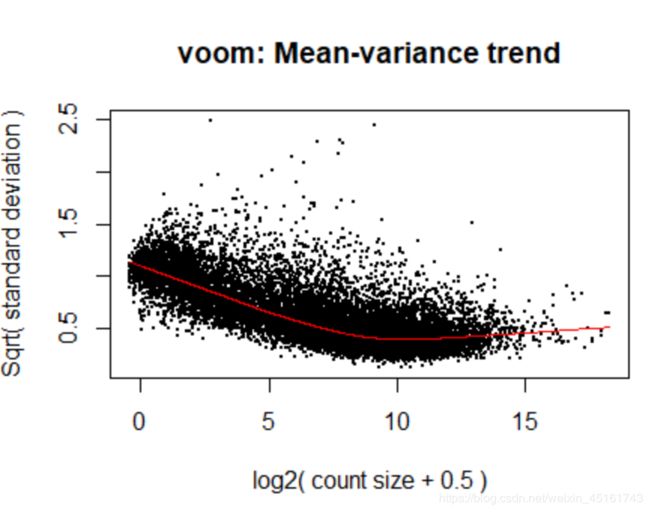
ps:
1)方差来源: 测序实验中的技术差异和不同细胞类型的重复样本之间的生物学差异的结合
2)voom图趋势: 会显示一个在均值与方差之间递减的趋势,生物学差异高的实验通常会有更平坦的趋势,其方差值在高表达处稳定;生物学差异低的实验更倾向于急剧下降的趋势
3)检查过滤充不充分: 如果对于低表达基因的过滤不够充分,在图上表达低的一端,受到非常低的表达计数的影响,可以观察到方差水平的下降。如果观察到这种情况,应当回到最初的过滤步骤并提高用于该数据集的表达阈值
4)voom对象存储内容: v$genes=dge$genes,v$targets=dge$samples,v$E类似于dge$counts,以及精确权重矩阵v$weights,设计矩阵v$design
5)voom作用: 调整均值与方差的关系,适用于变异大的数据
3. 建模及结果
使用的是voom归一化得到的数据
- 计算结果
#拟合线性模型
fit <- lmFit(v, design)
#针对给定的对比计算估计系数和标准误差
fit2 <- contrasts.fit(fit, cont.matrix)
#计算出t统计量,F统计量和差异表达倍数的对数
fit2 <- eBayes(fit2)
- 提取差异基因
- 方法1: 直接卡阈值
allDEG <- topTable(fit2, coef = 'trt-untrt', n = Inf)
allDEG <- na.omit(allDEG)
padj = 0.05
foldChange= 1
diff_signif = allDEG[(allDEG$adj.P.Val < padj & abs(allDEG$logFC)>foldChange),]
diff_signif = diff_signif[order(diff_signif$logFC),]
save(diff_signif, file = 'limma_diff.Rdata')
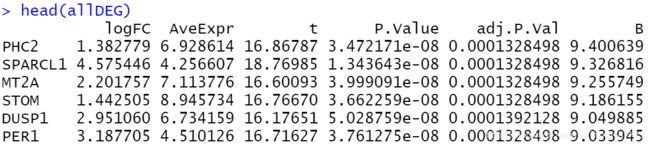

- 方法2: 使用decideTests函数
#查看差异基因数目
summary(decideTests(fit2,lfc=1,p.value = 0.05))

decideTests得出的结果和直接卡阈值得出的结果有出入,可能是因为计算p.adj的方法不同
#提取
allDEG <- topTable(fit2, coef = 'trt-untrt', n = Inf)
allDEG <- na.omit(allDEG)
dt <- decideTests(fit2)
de.common <- which(dt[,1]!=0)
diff<-allDEG[de.common,]

4.检查
- 从上到下检查单个DE基因
#将基因按照校正p值从小到大排列输出所有结果
df <- topTreat(fit2, coef=1, n=Inf)

- 基因集检验
基因集获取:http://bioinf.wehi.edu.au/software/MSigDB/
if (!require("org.Hs.eg.db", quietly = TRUE)) #注释基因
BiocManager::install("org.Hs.eg.db")
if (!require("clusterProfiler", quietly = TRUE)) #ID转换
BiocManager::install("clusterProfiler")
load("human_c2_v5p2.rdata")
#使用的是voom归一化的数据
geneid<-bitr(rownames(v), fromType='SYMBOL', toType='ENTREZID',OrgDb='org.Hs.eg.db', drop = TRUE)
idx <- ids2indices(Hs.c2,id=geneid$ENTREZID) #匹配基因集
cam<- camera(v,idx,design,contrast=cont.matrix)

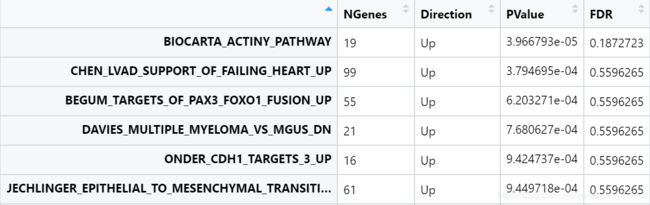
ps:
1) 原理: 通过比较假设检验来评估一个给定基因集中的基因是否相对于不在集内的基因而言在差异表达基因的排序中更靠前。 再通过基因间相关性和基因集的规模得到方差膨胀因子,并使用它调整基因集检验统计值的方差后,将会返回根据多重假设检验进行了校正的p值
2)输入: limma的线性模型框架,设计矩阵和对比矩阵(如果有的话),且在测试的过程中会使用来自voom的观测水平权重
3)应用: 适合检验基因集的大型数据库并观察其中哪些相对于其他的在排序上位次更高,适用于搜寻具有意义的基因集(mroast测试的是已经确定有意义的基因集的显著性)