call peak的另一个软件-----SICER
说到ChIP-Seq数据的call peak,第一个想到的软件肯定是大名鼎鼎的MACS2。MACS2比较适合call narrow peak,当然broad peak也是可以的。但是我课题的数据却不适用于这个软件,无奈之下我又尝试了另一个软件SICER。SICER适用于找出那些diffuse/broad peak,通过识别空间信号簇来寻找ChIP-enriched信号。(下面是原理,其实一知半解,文献是关于算法的,原谅我数学统计知识真的不好)
一、Complete SICER flowchart
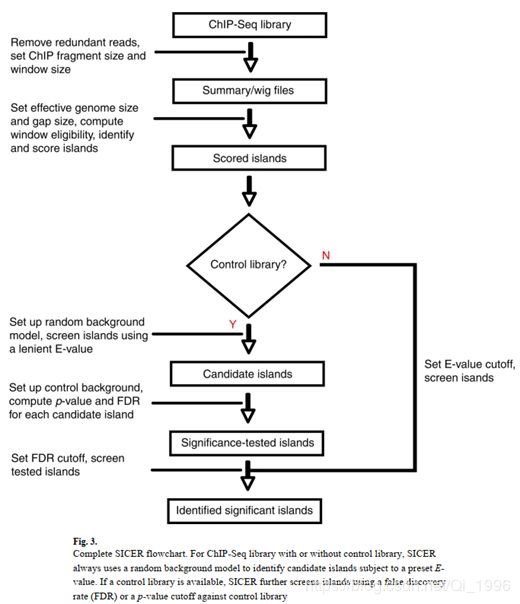
二、原理
下面是对island的定义:
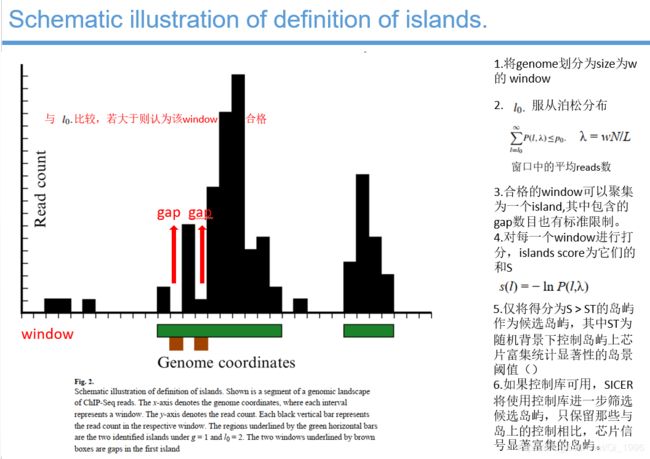
附上网盘资源(包含文献和使用教程,有问题的宝宝自己看文献吧,捂脸)
链接:https://pan.baidu.com/s/1UqivCVm935JarAzXfDmqJQ
提取码:p5gm
复制这段内容后打开百度网盘手机App,操作更方便哦
三、安装
1.下载安装包
wget https://home.gwu.edu/~wpeng/SICER_V1.1.tgz
2.解压
tar xvzf SICER_V1.1.tgz
3.vim SICER.sh SICER-rb.sh
将第一行按如下修改
PATHTO=/home/data/SICER1.1
to
PATHTO=/mydir/SICER_V1.1
4.vim SICER-df.sh and SICER-df-rb.sh
将第一行按如下修改
SICER=/mydir/SICER_V1.1/SICER
安装成功后可以看到文件夹里有四个shell脚本文件:SICER.sh,SICER-rb.sh,SICER-df-rb.sh,SICER-df.sh。
四、使用
如果有ChIP library & control library ,就用主脚本SICER.sh;
如果没有control library,可以用SICER-rb.sh替代SICER.sh.
另外两个是用来找差异部分,还没使用过。
1.Command:
SICER.sh takes 11 ordered command line parameters. The general command structure is
$ sh /mydir/SICER_V1.1/SICER/SICER.sh [Input directory] [ChIP file] [Control file][Output directory] [Species] [Redundancy threshold] [Window size] [Fragment size] [Effective genome fraction] [Gap size] [FDR]
具体参数解释如下:
[Input directory]:输入文件所在目录
[Control file]:输入文件名
[Output directory]:输出文件所在目录
[Species]:mm8, mm9, rn4, hg18, hg19, sacCer1, dm2, dm3, pombe, and tair8
[Redundancy threshold]:保留用于分析的冗余读数。一般默认为1.
[Window size]:芯片与控制库比较时窗口的宽度(单位为bp)。根据需要自行修改,文献示例为200bp。如果是very broad peak 可以适当放大。
[Fragment size]:芯片碎片的平均大小(单位为bp)。在示例中,默认推荐值是150。该参数用于将读取的芯片分配到DNA片段的中心。
[Effective genome fraction]:在示例中,是0.8。一般也默认0.8。
[Gap size]:在SICER中允许的间隙大小(以bp为单位)。在示例中,它是600 bp(等于3个窗口),推荐用于扩展组蛋白修饰标记,如H3K27me3。此参数必须是窗口大小的倍数。
[FDR]:在识别具有统计意义的岛屿时所需的错误发现率。在这个例子中,FDR = 0.1%。
2.输出文件
翻译不动了,大家凑合看一下英文说明吧。在所有这些文件中,ES_H3K27me3-W200-G600-islands-summary-FDR1e-3和ES_H3K27me3-W200-G600-FDR1e-3-island.bed这两种是最重要的,可以用于进一步分析。

8月去北京开会有幸遇到了SICER软件的开发者臧充之老师,交流之后得知老师组建实验室后更新了SICERv2.0。安装之后运行发现有三个比较明显的变化:
1.输入文件不再局限于bed格式,bam文件也是可以的。
2.命令格式也发生了变化,如果用到v2版本可以去实验室主页看下
3.运行速度大大提升,输出文件去掉了一些我觉得比较冗余的
实验室SICER2网址:
https://zanglab.github.io/SICER2/#sicer2