前面已经通过TCGA官方API下载了肝癌373个cases的临床信息(如days_to_death,state等),还下载了425个样本的类型(是tumor还是normal)数据,这次我们从这两个文件提取关键信息并进行整合.
首先提取../cases_373_diagnoese.tsv文件中的信息,这个使用R脚本比较方便,R脚本如下,保存为subCols.R文件.
#########################
# 2017.5.28
rm(list=ls())
# 读入参数:
args<-commandArgs(trailingOnly = TRUE)
# 第一个参数是文件路径:
files_path<-args[1]
# 第二个参数是输出路径:
out_path<-args[2]
# 使用向量保存传入的数组:
subCols <- as.vector(args[3:length(args)])
# 读入文件:
files_readed<-read.table(files_path,stringsAsFactors = F,sep="\t",header = T)
# 写入结果:
write.table(files_readed[,subCols],file=out_path,append = F,quote = F,sep="\t",col.names = T,row.names = F)
然后在shell下直接调用subCols.R,第一个参数是cases_373_diagnoese.tsv文件,第二个参数是输出文件名,第三列是要提取列的shell下的数组,shell下的命令如下:
# 提取../cases_373_diagnoese.tsv部分信息,包括以下列:
cases_373_subCol=(case_id diagnoses_0_days_to_last_follow_up diagnoses_0_days_to_birth diagnoses_0_tumor_stage diagnoses_0_age_at_diagnosis disease_type diagnoses_0_state primary_site diagnoses_0_days_to_death diagnoses_0_morphology state diagnoses_0_vital_status updated_datetime )
#用R脚本 ../subCols.R,第一个参数是输入文件,第二个参数是输出文件,最后一个是上面定义的数组:
Rscript ../subCols.R ../cases_373_diagnoese.tsv ../cases_373_diagnoese_sub.tsv ${cases_373_subCol[*]}
结果文件是cases_373_diagnoese_sub.tsv:
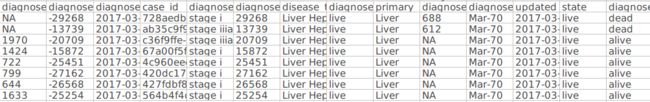
cases_373_diagnoese.tsv提取列后的文件.png
同理使用上面的脚本提取../files_425_diagnoese.tsv文件中的部分列:
# 提取../files_425_diagnoese.tsv部分信息,包括以下列:
files_425_subCol=(cases_0_case_id file_id cases_0_samples_0_sample_type)
#用R脚本 ../subCols.R,第一个参数是输入文件,第二个参数是输出文件,最后一个是上面定义的数组:
Rscript ../subCols.R ../files_425_diagnoese.tsv ../files_425_diagnoese_sub.tsv ${files_425_subCol[*]}
结果保留为files_425_diagnoese_sub.tsv ,其实提取的有效信息只有一列,也就是sample_type,也就是425个样本中哪些是正常组织的,哪些是肿瘤的.
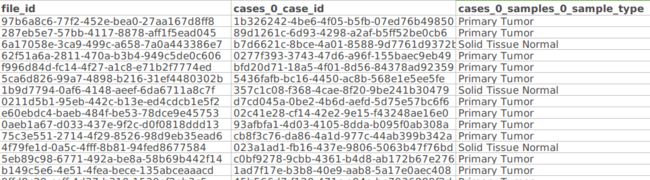
融合两个提取的文件获得425个样本的样本类型以及所对应case的days_to_death等信息.
# 整合样本类型和cases临床信息数据:
awk 'BEGIN{OFS="\t"}{
if(NR==FNR){a[$1]=$0}
if(NR>FNR&&FNR==1){print $0,a["case_id"]} #case_id列名不同,中叫case_id,425中叫case0_case_id,所以单独矫正.
if(NR>FNR&&FNR>1){print $0,a[$1]|"sort -k 3,3r -k 1,1"} #按照
}' ../cases_373_diagnoese_sub.tsv ../files_425_diagnoese_sub.tsv>../cases_files_merged.tsv
cases_files_merged.tsv就是得到的整合后的临床数据文件.但在这个表中,并没有明确给出生存时间.并且对于已经dead的人,生存时间是diagnoses_0_days_to_death, 然而还alive的人是没有这个时间的,所以对于alive人来说,生存时间是diagnoses_0_days_to_last_follow_up.我们用个R小脚本得到包含生存时间的最终的临床数据文件.
将下面的脚本存为survival_deal.R文件.
rm(list=ls())
# 获得参数数组:
args<-commandArgs(trailingOnly = TRUE)
# 输入文件路径:
files_path<-args[1]
# 输出文件路径:
out_path<-args[2]
# 读入文件:
files_readed<-read.table(files_path,stringsAsFactors = F,sep="\t",header = T)
# dead人的survival time 等于diagnoses_0_days_to_death
files_readed[files_readed$diagnoses_0_vital_status=="dead","survival_time"]=files_readed[files_readed$diagnoses_0_vital_status=="dead","diagnoses_0_days_to_death"]
# alive人的survival time 等于diagnoses_0_days_to_last_follow_up
files_readed[files_readed$diagnoses_0_vital_status=="alive","survival_time"]=files_readed[files_readed$diagnoses_0_vital_status=="alive","diagnoses_0_days_to_last_follow_up"]
#写入文件:
write.table(files_readed,file=out_path,append = F,quote = F,sep="\t",col.names = T,row.names = F)
运行survival_deal.R需要两个参数,第一个是输入文件的路径,也就是刚生成的文件.第二个是输出文件的路径.然后运行下面的代码:
# 运行R脚本:
Rscript ../survival_deal.R ../cases_files_merged.tsv ../cases_files_merged_survival.tsv
结果文件../cases_files_merged_survival.tsv包含了生存时间数据:
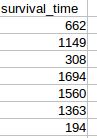
最后一列生存时间数据
TCGA表达谱及临床数据下载和处理到这里就基本结束了,下期开始使用这些数据进行分析.
更多原创精彩内容敬请关注生信杂谈:
