个人基因组比对及其变异分析
这里用到生物学软件有
1.bwa(主要用途a.建立参考基因组索引文件的。b.进行比对)类似的建立索引的软件有bowtie,hisat
http://sourceforge.net/projects/bio-bwa/files/
2.samtools(主要用途a.排序b.合并c.建立基因组index)
http://sourceforge.net/projects/samtools/files/samtools/
该软件需要编译安装
3.bcftools(主要用途:view命令来进行SNP和Indel calling,它是samtools中附加的软件)
4.snpEff (主要用途:a.添加标签b.标记dbSNP的ID号)
5.freebayes(主要用途:添加一些标签)
(以上软件需要配置环境变量,添加方法见这里)
一、基因组下载
下载参考基因组hg19,所在的网站如下:
http://hgdownload.cse.ucsc.edu/goldenPath/hg19/bigZips/chromFa.tar.gz
下载至~/reference/genome/hg19/目录
下载过后解压,cat合并成hg19.fa一个文件
下载需要比对的个人基因组,这里我拿到的是韩国人个人基因组计划中编号为KPGP-00001的基因组,所在ftp站点:
ftp://biodisk.org/Release/KPGP/KPGP_Data_2016_Release_Candidate/WGS/KPGP-00001
下载至~/Data/KPGP-00001目录下
下载过后注意md5校验,md5sum + 文件名;
二、bwa软件建立索引
新建路径~/reference/index/bwa/,用以存放参考基因组的索引。
bwa index -a(STR BWT construction algorithm) bwtsw -p(STR prefix of the index [same as fasta name]) ~/reference/index/bwa/hg19 ~/reference/genome/hg19/hg19.fa 1>hg19.bwa.index.log 2>&1
因为运行时间较长,此命令最好在后台运行,(nohup time,加上time可以看见运行时间)
三、将fastq文件比对到hg19参考基因组
bwa mem(PE150测序比对模式) -t (INT number of threads) -M(mark shorter split hits as secondary 选择上一步建立好的参考基因组索引) ~/reference/index/bwa/hg19 KPGP-00001_R1.fq.gz KPGP-00001_R2.fq.gz 1>KPGP-00001.sam 2>KPGP-00001.bwa.log
若有多个fq文件,需要加for流程控制,得到多个sam文件。此命令在后台运行。
至此,我们用已经比对得到了sam格式比对结果文件,从sam文件中我们看到每条reads在基因组及染色体的位置
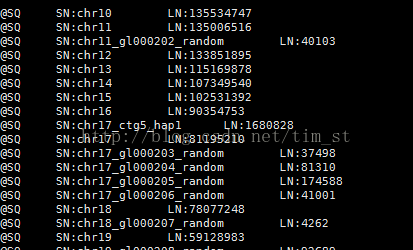
图一
若图片不清晰,可放大网页查看。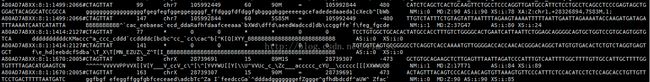
图二
sam文件共12列,比对软件不同会有不同列数。每列的意义如下:
第一列:read name,read的名字通常包括测序平台等信息;
第二列:sum of flags,每个flag用数字来表示,分别为:
1 read是pair中的一条(read表示本条read,mate表示pair中的另一条read)
2 pair一正一负完美的比对上
4 这条read没有比对上
8 mate没有比对上
16 这条read反向比对
32 mate反向比对
64 这条read是read1
128 这条read是read2
256 第二次比对
512 比对质量不合格
1024 read是PCR或光学副本产生
2048 辅助比对结果
通过这个和可以直接推断出匹配的情况。假如说标记不是以上列举出的数字,比如说83=(64+16+2+1),就是这几种情况值和。
第三列:RNAM,reference sequence name,实际上就是比对到参考序列上的染色体号。若是无法比对,则是*;
第四列:position,read比对到参考序列上,第一个碱基所在的位置。若是无法比对,则是0;
第五列:Mapping quality,比对的质量分数,越高说明该read比对到参考基因组上的位置越唯一;
第六列:CIGAR值,read比对的具体情况,
“M”表示 match或 mismatch;
“I”表示 insert;
“D”表示 deletion;
“N”表示 skipped(跳过这段区域);
“S”表示 soft clipping(被剪切的序列存在于序列中);
“H”表示 hard clipping(被剪切的序列不存在于序列中);
“P”表示 padding;
“=”表示 match;
“X”表示 mismatch(错配,位置是一一对应的);
第七列:MRNM(chr),mate的reference sequence name,实际上就是mate比对到的染色体号,若是没有mate,则是*;
第八列:mate position,mate比对到参考序列上的第一个碱基位置,若无mate,则为0;
第九列:ISIZE,Inferred fragment size.详见Illumina中paired end sequencing 和 mate pair sequencing,是负数,推测应该是两条read之间的间隔(待查证),若无mate则为0;
第十列:Sequence,就是read的碱基序列,如果是比对到互补链上则是reverse completed
第十一列:ASCII,read质量的ASCII编码。
第十二列之后:Optional fields,可选的区域
| Tag | Type | Description |
| X? | ? | Reserved fields for end users (together with Y? and Z? ) |
| AM | i | The smallest template-independent mapping quality of fragments in the rest |
| AS | i | Alignment score generated by aligner |
| BQ | Z | Offset to base alignment quality (BAQ),of the same length as the read sequence.At the i-th read base,BAQi= Qi- (BQi- 64) where Qi is the i-th base quality. |
| CM | i | Edit distance between the color sequence and the color reference (see also NM ) |
| CQ | Z | Color read quality on the original strand of the read.Same encoding as QUAL; same length as CS. |
| CS | Z | Color read sequence on the original strand of the read.The primer base must be included. |
| E2 | Z | The 2nd most likely base calls.Same encoding and same length as QUAL. |
| FI | i | The index of fragment in the template. |
| FS | Z | Fragment suffix. |
| LB | Z | Library.Value to be consistent with the header RG-LB tag if @RG is present. |
| HO | i | Number of perfect hits |
| H1 | i | Number of 1-difference hits (see also NM) |
| H2 | i | Number of 2-difference hits |
| HI | i | Query hit index,indicating the alignment record is the i-th one stored in SAM |
| IH | i | Number of stored alignments in SAM that contains the query in the current record |
| MD | Z | String for mismatching positions.Regex: [0-9]+( ([ACGTN] |\^ [ACGTN]+) [0-9]+)* |
| MQ | i | Mapping quality of the mate/next fragment |
| NH | i | Number of reported alignments that contains the query in the current record |
| NM | i | Edit distance to the reference,including ambiguous bases but excluding clipping |
| OQ | Z | Original base quality (usually before recalibrat ion).Same encoding as QUAL. |
| OP | i | Original mapping position (usually before realignment ) |
| OC | Z | Original CIGAR (usually before realignment) |
| PG | Z | Program.Value matches the header PG-ID tag if @PG is present. |
| PQ | i | Phred likelihood of the template,conditional on both the mapping being correct |
| PU | Z | Plat form unit.Value to be consistent with the header RG-PU tag if CRG is present. |
| Q2 | Z | Phred quality of the mate/next fragment.Same encoding as QUAL. |
| R2 | Z | Sequence of the mate/next fragment in the template. |
| RG | Z | Read group.Value matches the header RG-ID tag if QRG is present in the header. |
| SM | i | Template-independent mapping quality |
| TC | i | The number of fragments in the template. |
| U2 | Z | Phred probility of the 2nd call being wrong conditional on the best being wrong.The same encoding as QUAL. |
| UQ | i | Phred likelihood of the fragment,conditional on the mapping being correct |
samtools sort -o *_sorted.bam *.sam 即可直接生成排序过后的bam文件,若有多个sam文件,需加for流程控制,生成多个bam文件,然后samtools merge *_merge.bam *_sorted.bam 将所有排序过的bam文件合并成一个bam文件。
最后,将合并好的bam文件再次排序:samtools sort -O(制定输入格式为bam) bam -o *sorted_merge.bam *_merge.bam
接下来建立索引,建立索引前,必须对bam文件进行默认情况下的排序后,才能进行index。否则会报错。
samtools index 后会产生.bai格式文件。现在你可以使用samtools flagstat 命令给出BAM文件的比对结果,生成txt文件。(txt文件每一项意义见http://blog.csdn.net/tim_st/article/details/77941969)
至此,我们已经大致处理了一下拿到手中的数据,得到了比对结果(sam文件和bam文件)。当然,我们的得到的比对结果太过粗糙,因为其中包含比对的数据和左右端测序数据比对到不同的染色体的PE reads。不过问题不大,只是结果有些粗糙罢了。接下来我们要得到vcf格式的基因比对结果,call出varitation.
四、用bcftools来call varitation。
在call vcf文件之前,要将得到的bam文件转为mpileup文件:
samtools mpileup -ugf hg19.fa(此基因组要与参考基因组相同,此处是hg19,有些是GRCh37、hg38等) *sorted_merge.bam |bcftools call -vmO z -o *.bcftools.vcf.gz
这里的参数可以查看samtools和bcftools帮助文档。
漫长的等待……就能看到vcf文件了。现在,我们已经得到了碱基有哪里不同的信息,从下图也可以看出。vcf文件信息代表意义
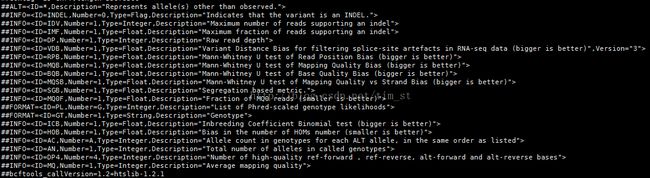
图三
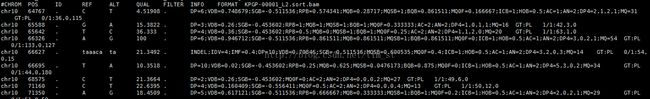
图四
然而,仅仅vcf文件是不能够确定是否发生了基因突变的,只能知道哪个碱基突变了。vcf格式文件务必能大致看得懂,因为接下来我们要用一些软件再向vcf文件中添加一些标签(tag)。用SnpSift添加variation标签;freebayes添加标签。

图五
五、把突变文件注释到dbsnp数据库
公用数据库注释找到具体的基因突变信息。首先下载dbSNP数据库ftp://ftp.ncbi.nih.gov/snp/organisms/
找到human开头的有几个版本这里选择的human_9606_b150_GRCh37p13。这里存在一些问题:我不知道这些版本有什么区别0.0 我下载的是这个版本的。欢迎各位看官指点。
![]()
图六
java -jar SnpSift.jar varType *.vcf > *.varType 稍等就能得到加了标签的vcf文件,如图五。对比图四图五图七,在INFO那列会多出一些标签。

图七
最后我们用dbsnp数据库注释,java -jar SnpSift.jar annotate All_20161121.vcf.gz *.vcf > *.dbsnp.vcf
注释过后文件如图八

图八
我们加过标签的文件,和经过注释的文件的头文件为各个标签的解释信息,注意看一下。
最后一步
最后就是使用脚本语言提取出vcf文件的标签了,这里不再一一举例,各位想要什么信息就自己提取吧!这里小编学习的是shell脚本和perl脚本。
祝顺利!望各位多多指点!