- Microsoft Defender SmartScreen 阻止了无法识别的应用启动。运行此应用可能会导致你的电脑存在风险。
开心呆哥
microsoft
你提到的情况可能是由于Windows的安全策略导致的。当你运行批处理文件(.bat)时,Windows可能会弹出一次提示,询问是否允许该文件执行。这是为了确保用户不会意外地运行潜在的恶意脚本。有几种方法可以解决这个问题:解锁文件:在文件资源管理器中,找到你的.bat文件。右键单击文件,选择"属性"。在"常规"选项卡中,如果有一个"解锁"复选框,请勾选它。确定并尝试重新运行文件。以管理员身份运行:右
- 解决“Python中 pip不是内部或外部命令,也不是可运行的程序或批处理文件”的方法。
གཡུ །
Python常规问题pythonpip机器学习自然语言处理
解决‘Python中pip不是内部或外部命令,也不是可运行的程序或批处理文件。’的方法1、pip是什么?pip是一个以Python计算机程序语言写成的软件包管理系统,他可以安装和管理软件包,另外不少的软件包也可以在“Python软件包索引”中找到。它可以通过cmd(命令提示符)非常方便地下载和管理Python第三方库,比如,Python爬虫中常见的requests库等。但是我们在使用cmd运行pi
- ‘pip‘ 不是内部或外部命令,也不是可运行的程序或批处理文件。
逆天小北鼻
pythonpip
因为python13不支持pipinstallcx_Oracle,卸载了python13,重新安装python10,导致cmd命令不识别pip,和python10,(;´༎ຶД༎ຶ`)记录一种临时方案如果你的命令行界面(cmd)不识别pip命令,这通常意味着pip的可执行文件路径没有被添加到系统的环境变量中,或者环境变量的设置没有正确生效。重新检查pip的安装路径在命令行中运行以下Python代码
- pip‘ 不是内部或外部命令,也不是可运行的程序 或批处理文件。
knighthood2001
pip
重新设置一下环境变量。注意,这里后面没有斜杠我之前就是因为环境变量中,这两行最后都有斜杠,导致提示pip‘不是内部或外部命令,也不是可运行的程序或批处理文件。
- 【Java】读取超大文件的时候,如何避免出现OOM
七七r
javapython开发语言
读取超大文件的时候,如何避免出现OOM需求背景如下:从文件中读取数据并经过业务处理后存储到数据库中,同时避免出现OOM(OutofMemory)1、使用分批处理文件数据将文件数据分批读取,每次只处理一部分数据,避免一次性将整个文件加载到内存中。可以使用Java的BufferedReader和InputStreamReader类来实现分批读取文件数据。importjava.io.*;importja
- ‘pip’ 不是内部或外部命令,也不是可运行的程序或批处理文件解决方案。
雪山之巅GGG
Pythonpythonpip
问题描述:不能运行pip命令在Pycharm的Terminal的命令框中不能运行pip命令,提示’pip’不是内部或外部命令,也不是可运行的程序或批处理文件。'pip'不是内部或外部命令,也不是可运行的程序或批处理文件。原因分析及解决方案:1:检查pip在CMD命令窗口中是否能够运行:如果不能运行,检查环境变量中是否添加了pip的路径。[添加路径参考:https://zhidao.baidu.co
- maven安装配置之后mvn命令仍然无效的解决办法
ZhangYuanhao_1995
maven
问题描述:配置了maven的环境变量,用mvn-v提示不是内部或外部命令,也不是可运行的程序或批处理文件。解决办法:1.在系统变量Path那添加%M2_HOME%\bin路径,2.在用户变量的Path那也添加%M2_HOME%\bin。之后就能正常使用mvn了。
- 佳佳mpg格式转换器免费版 v12.4.0.0
ldy721224
多媒体类视频转换MPGmpg格式转换器
点击下载来源:佳佳mpg格式转换器免费版v12.4.0.0佳佳MPG格式转换器是一款功能强大的MPG、MPEG-2视频格式转换器,mpg格式转换器它可以带给您超高速和超高质量视频转换体验。可以转换成DivX,XviD,AVI,WMV,MPG,MPEG,MPG,MP4,MKV等流行的视频格式。以便用户刻录DVD、VCD、SVCD或者用会声会影进行二次编辑。并且支持支持批处理文件的转换。本款mpg格式
- 2021-08-23 学习小组Day3笔记--动感光波
e4fdf8628246
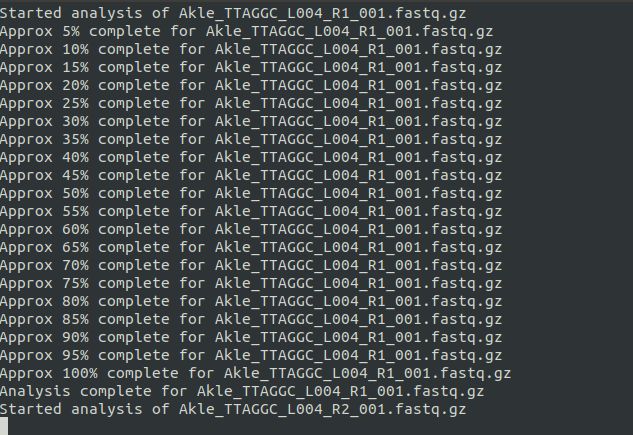
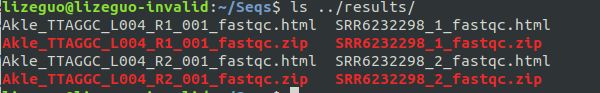
Linux入门--软件安装首先安装conda,这是个相当于linux系统下的应用商店的存在。日常生信用miniconda即可。安装好miniconda后,下载fastqc软件。miniconda安装网页搜索“miniconda清华”镜像(清华的conda镜像网站)首先,查询自己的电脑是多少位的:uname-a结果是64位,然后,在网页搜“miniconda清华”,找到Linux最新版本的(late
- npm run dev运行出现NODE_OPTIONS=--max_old_space_size=4096 vite --mode dev --host?
舒一笑
Vuenpm前端node.js
问题描述PSE:\AWorkDataease\DataEase\core\core-frontend>
[email protected]_OPTIONS=–max_old_space_size=4096vite--modedev--host0.0.0.0‘NODE_OPTIONS’不是内部或外部命令,也不是可运行的程序或批处理文件。解决方案遇到'NODE_OPTIONS'
- Win10_cmd下提示:‘xxx’不是内部或外部命令,也不是可运行的程序 或批处理文件
笑胖仔
一、windows下命令行查看ip:win+R,调出【运行】,输入cmd,回车打开终端,输入ipconfig,查看成功:二、可能出现的问题:‘ping’不是内部或外部命令,也不是可运行的程序或批处理文件。‘ipconfig’不是内部或外部命令,也不是可运行的程序或批处理文件。解决:1、控制面板-系统:2、高级系统设置-环境变量:3、找到系统变量中Path:4、双击打开Path,点新建:5、将该路径
- bat 定时收缩sqlserver2017
三希
sqlserver数据库oracle
如果你希望使用批处理(.bat)文件来定时收缩SQLServer的数据库,你可以编写一个脚本来执行这个任务。但首先,需要注意的是,定期收缩数据库通常不是一个好的做法,因为它可能会对性能产生负面影响,并可能导致数据库碎片化。然而,如果你确实需要这样做,以下是一个简单的示例,展示了如何使用SQLCMD工具在批处理文件中执行收缩数据库的操作:@echooffset"DBNAME=YourDatabase
- 批量为文件重命名,不会这招就可惜了
新精英充电站
很多职场人士都会遇到需要修改文件名的情况。如果文件不多,选中文件,按F2就可以快速重命名。但是文件数量太多,例如有100个文件要重命名,一个一个地来,得多久呀。其实文件重命名也有巧秒的方法,其原理是:用dos下的批处理文件bat文件来为文件重命名。而bat文件可以修改txt记事本文件而来。但是还有一个关键点,需要在bat文件写清楚文件的原名和修改后的名称。所以还要借助Excel快速提取原文件名称及
- python找不到requests模块(No module named requests)解决方案
橘向阳
python
python找不到requests模块(Nomodulenamedrequests)解决方案报错原因:没有安装requests的模块解决方法:安装requests的模块即可\Python38>pipinstallrequests'pip'不是内部或外部命令,也不是可运行的程序或批处理文件报错:‘pip’不是内部或外部命令,也不是可运行的程序或批处理文件解决方法:找到python安装下的目录,进入到
- pc端微信多开。
L762455597
学习微信
目录前言一、bat文件代码及解读二、实践总结前言学习需要多开微信,但是pc端只允许单开微信,了解到可以通过批处理文件来实现多开。一、bat文件代码及解读@echooff//关闭命令显示正在执行的批处理命令及执行的结果taskkill/F/IM微信.exe//关闭正在执行的微信进程start"""微信.exe位置"//执行微信启动文件start"""微信.exe位置"//同理,需要多开几个就开几行二
- ‘vue-cli-service‘ 不是内部或外部命令,也不是可运行的程序
舒一笑
Vuevue.js前端javascript
遇到'vue-cli-service'不是内部或外部命令,也不是可运行的程序或批处理文件。的错误时,通常意味着VueCLI没有被正确安装或配置在项目中。这可能是因为node_modules目录缺失了必要的包,或者局部安装的VueCLI没有被正确设置到系统的PATH环境变量中。下面是几个步骤来解决这个问题:确保VueCLI已安装:首先,确保你已经全局安装了VueCLI。可以通过下面的命令来安装:np
- bat通过ssh增量同步文件夹
三希
ssh运维
要通过SSH使用批处理文件(.bat)进行文件夹的增量同步,你可以使用rsync命令,这是一个非常强大的文件同步工具,它支持增量同步。以下是一个简单的批处理脚本示例,该脚本使用SSH连接到一个远程服务器,并使用rsync进行增量同步。首先,确保你的本地计算机上安装了SSH客户端(如OpenSSH)和rsync。在Windows上,你可以使用像GitBash、Cygwin、WindowsSubsys
- Windows路径含有带空格的目录/文件名的处理
中年陀罗
今天在学习打包工具,的时候遇到了一个问题:return"C:/ProgramFiles/Unity/Hub/Editor/2019.3.10f1/Editor/Unity.exe"返回代用一个的应用程序的时候报错:C:/Program'不是内部或外部命令,也不是可运行的程序或批处理文件。在网上查了下原因:命令行被空格符截断了。解决办法:1使用双引号:return"C:/\"ProgramFiles
- 无法打开项目文件。 无法找到 .NET SDK。请检查确保已安装此项且 global.json 中指定的版本(如有)与所安装的版本相匹配
%xiao Q
C#c#
问题:如果在运行c#代码出现以下情况:无法打开项目文件。无法找到.NETSDK。请检查确保已安装此项且global.json中指定的版本(如有)与所安装的版本相匹配解决方案:我们可以先看一下是否安装.NETCore,打开cmd输入dotnet回车运行,如果出现’dotne’不是内部或外部命令,也不是可运行的程序或批处理文件。则是没有安装.NETCore,安装.NETCore即可。安装地址:http
- WebStorm中在Terminal,npm命令不能使用: ‘'npm' 不是内部或外部命令,也不是可运行的程序 或批处理文件问题
技术小菜鸟
点击File->Settings->Tools->Terminal,点击EnvironmentVariables如图示位置image.pngimage.png勾选includesystemenvironmentvariables
- 2022-11-26
fb6697f26c1b
生信记录11.fastqc路径/root/anaconda3/bin/FastQC/执行./root/anaconda3/bin/FastQC/fastqc结果/root/sratoolkit.3.0.1-centos_linux64/bin2.prefetch路径/root/sratoolkit.3.0.1-centos_linux64/bin执行./prefetch结果/root/sratoo
- Java 学习和实践笔记(3)
复业思维20240108
学习笔记
安装和配置成功:运行第一个程序时出现这个错误:'javac'不是内部或外部命令,也不是可运行的程序或批处理文件。找到这篇文章看了下:'javac'不是内部或外部命令,也不是可运行的程序或批处理文件。_javac'不是内部或外部命令,也不是可运行的程序或批处理文件。-CSDN博客仅仅关掉cmd再重启,就解决了问题:
- Vue前端框架--Vue工程项目问题总结{脚手架 Vue-cli}
鲲鹏猿
前端框架vue.jsarcgis
Vue脚手架部署问题总结我所遇到的一共两大问题只有先执行npminstall之后才能runserve否则会报错vue-cli-serve不是内部或者外部的命令,也不是可运行的程序或者批处理文件的错误1.运行npminstall会报错2.运行npmrunserve报错nodejs官网为https://nodejs.org/en/选择推荐用户最多的使用版本关于第一次下载nodejs需要注意最好是按照默
- npm run dev 疑难解答
鲁正杰
npm前端node.js
在命令行下执行命令:npmrundev以下几个是典型错误,记录解决办法分享给大家:'vite'不是内部或外部命令,也不是可运行的程序或批处理文件。npminstall-gviteError:Cannotfindmodule'@vitejs/plugin-vue'npmuninstall@vitejs/plugin-vuenpminstall-gcnpm--registry=https://regi
- bioinfo100-第9题-FastQC报告中的duplicate
RachaelRiggs
duplicate问题zhn去除duplicate可以这样理解:去除“假重复”(人为造成的重复序列方面的bias)保留“真重复”(天然存在的重复序列)。第9题读懂FastQC报告中的duplicate问题本周我们预计会把前10个问题提出来,结束我们的测序原理与FastQC部分。今天我们来详细聊聊duplicate问题。duplicate的产生主要是因为Illumina建库的过程中,一般会需要使用P
- ‘vue-cli-service‘ 不是内部或外部命令,也不是可运行的程序 或批处理文件。这个问题如何解决?
舒一笑
Vuevue.js前端javascript
这个错误信息'vue-cli-service'不是内部或外部命令,也不是可运行的程序或批处理文件表示vue-cli-service命令在你的系统上未被识别。这通常是因为VueCLI没有被正确安装或其路径没有被加入到系统的环境变量中。以下是几个解决这个问题的步骤:确认VueCLI是否已安装:打开命令行,输入vue--version。如果VueCLI已安装,这个命令会显示其版本号。如果没有显示版本号或
- ‘vue-cli-service‘ 不是内部或外部命令,也不是可运行的程序或批处理文件。
石子君
问题及工具vuecil
运行vue项目发现报错,如图所示:‘vue-cli-service’不是内部或外部命令,也不是可运行的程序或批处理文件。解决办法:将项目里的“node_modules”文件夹删除(也可以不删),然后重新运行npminstall如果安装了淘宝镜像,可以运行cnpminstall。接着进行npmrunserve,大功告成!
- Vue项目运行时报错:‘vue-cli-service‘ 不是内部或外部命令,也不是可运行的程序 或批处理文件
蓝胖子的多啦A梦
vue.js前端javascript
报错原因及解决1.package.json文件中未定义依赖项@vue/cli-service,因此在npminstall之后并没有安装@vue/cli-service依赖;解决:项目目录下执行命令,npmi-D@vue/cli-service。2.第1步排查后,还是报同样的错;解决:尝试关闭编辑器和命令行面板,然后重启编辑器启动项目。此操作防止第1步安装依赖后环境变量不生效,导致vue-cli-s
- ‘vue-cli-service‘ 不是内部或外部命令,也不是可运行的程序 或批处理文件。
展望之客
vue.jsjavascript前端
报错:'vue-cli-service'不是内部或外部命令,也不是可运行的程序或批处理文件。解决办法:1.切换到项目目录,找到node_modules文件删除,如没有就执行如下命令2.执行命令:npminstall3.执行命令:npmrundev再次启动项目【项目执行】1、打开cmd,进入项目目录2、输入命令npmcleancache-f,清除npm缓存3、输入命令npminstall,重新安装依
- html用记事本打字显示问号,电脑记事本问号怎么办
亚赛大人
html用记事本打字显示问号
1.如何去除电脑桌面上的文件的问号1、通常是桌面上的图标变成左下方有问号标志。2、右键点击桌面→新建→文本文档。3、先复制以下命令,再在打开的记事本中粘贴一下。for/r.%%ain(.)do@ifexist"%%a\.svn"rd/s/q"%%a\.svn"4、然后再在工具栏点击文件→另存为。5、在文件名中输入:删除SVN信息.bat(注意扩展名更改)→保存。6、再双击该批处理文件即可解决问题。
- Algorithm
香水浓
javaAlgorithm
冒泡排序
public static void sort(Integer[] param) {
for (int i = param.length - 1; i > 0; i--) {
for (int j = 0; j < i; j++) {
int current = param[j];
int next = param[j + 1];
- mongoDB 复杂查询表达式
开窍的石头
mongodb
1:count
Pg: db.user.find().count();
统计多少条数据
2:不等于$ne
Pg: db.user.find({_id:{$ne:3}},{name:1,sex:1,_id:0});
查询id不等于3的数据。
3:大于$gt $gte(大于等于)
&n
- Jboss Java heap space异常解决方法, jboss OutOfMemoryError : PermGen space
0624chenhong
jvmjboss
转自
http://blog.csdn.net/zou274/article/details/5552630
解决办法:
window->preferences->java->installed jres->edit jre
把default vm arguments 的参数设为-Xms64m -Xmx512m
----------------
- 文件上传 下载 解析 相对路径
不懂事的小屁孩
文件上传
有点坑吧,弄这么一个简单的东西弄了一天多,身边还有大神指导着,网上各种百度着。
下面总结一下遇到的问题:
文件上传,在页面上传的时候,不要想着去操作绝对路径,浏览器会对客户端的信息进行保护,避免用户信息收到攻击。
在上传图片,或者文件时,使用form表单来操作。
前台通过form表单传输一个流到后台,而不是ajax传递参数到后台,代码如下:
<form action=&
- 怎么实现qq空间批量点赞
换个号韩国红果果
qq
纯粹为了好玩!!
逻辑很简单
1 打开浏览器console;输入以下代码。
先上添加赞的代码
var tools={};
//添加所有赞
function init(){
document.body.scrollTop=10000;
setTimeout(function(){document.body.scrollTop=0;},2000);//加
- 判断是否为中文
灵静志远
中文
方法一:
public class Zhidao {
public static void main(String args[]) {
String s = "sdf灭礌 kjl d{';\fdsjlk是";
int n=0;
for(int i=0; i<s.length(); i++) {
n = (int)s.charAt(i);
if((
- 一个电话面试后总结
a-john
面试
今天,接了一个电话面试,对于还是初学者的我来说,紧张了半天。
面试的问题分了层次,对于一类问题,由简到难。自己觉得回答不好的地方作了一下总结:
在谈到集合类的时候,举几个常用的集合类,想都没想,直接说了list,map。
然后对list和map分别举几个类型:
list方面:ArrayList,LinkedList。在谈到他们的区别时,愣住了
- MSSQL中Escape转义的使用
aijuans
MSSQL
IF OBJECT_ID('tempdb..#ABC') is not null
drop table tempdb..#ABC
create table #ABC
(
PATHNAME NVARCHAR(50)
)
insert into #ABC
SELECT N'/ABCDEFGHI'
UNION ALL SELECT N'/ABCDGAFGASASSDFA'
UNION ALL
- 一个简单的存储过程
asialee
mysql存储过程构造数据批量插入
今天要批量的生成一批测试数据,其中中间有部分数据是变化的,本来想写个程序来生成的,后来想到存储过程就可以搞定,所以随手写了一个,记录在此:
DELIMITER $$
DROP PROCEDURE IF EXISTS inse
- annot convert from HomeFragment_1 to Fragment
百合不是茶
android导包错误
创建了几个类继承Fragment, 需要将创建的类存储在ArrayList<Fragment>中; 出现不能将new 出来的对象放到队列中,原因很简单;
创建类时引入包是:import android.app.Fragment;
创建队列和对象时使用的包是:import android.support.v4.ap
- Weblogic10两种修改端口的方法
bijian1013
weblogic端口号配置管理config.xml
一.进入控制台进行修改 1.进入控制台: http://127.0.0.1:7001/console 2.展开左边树菜单 域结构->环境->服务器-->点击AdminServer(管理) &
- mysql 操作指令
征客丶
mysql
一、连接mysql
进入 mysql 的安装目录;
$ bin/mysql -p [host IP 如果是登录本地的mysql 可以不写 -p 直接 -u] -u [userName] -p
输入密码,回车,接连;
二、权限操作[如果你很了解mysql数据库后,你可以直接去修改系统表,然后用 mysql> flush privileges; 指令让权限生效]
1、赋权
mys
- 【Hive一】Hive入门
bit1129
hive
Hive安装与配置
Hive的运行需要依赖于Hadoop,因此需要首先安装Hadoop2.5.2,并且Hive的启动前需要首先启动Hadoop。
Hive安装和配置的步骤
1. 从如下地址下载Hive0.14.0
http://mirror.bit.edu.cn/apache/hive/
2.解压hive,在系统变
- ajax 三种提交请求的方法
BlueSkator
Ajaxjqery
1、ajax 提交请求
$.ajax({
type:"post",
url : "${ctx}/front/Hotel/getAllHotelByAjax.do",
dataType : "json",
success : function(result) {
try {
for(v
- mongodb开发环境下的搭建入门
braveCS
运维
linux下安装mongodb
1)官网下载mongodb-linux-x86_64-rhel62-3.0.4.gz
2)linux 解压
gzip -d mongodb-linux-x86_64-rhel62-3.0.4.gz;
mv mongodb-linux-x86_64-rhel62-3.0.4 mongodb-linux-x86_64-rhel62-
- 编程之美-最短摘要的生成
bylijinnan
java数据结构算法编程之美
import java.util.HashMap;
import java.util.Map;
import java.util.Map.Entry;
public class ShortestAbstract {
/**
* 编程之美 最短摘要的生成
* 扫描过程始终保持一个[pBegin,pEnd]的range,初始化确保[pBegin,pEnd]的ran
- json数据解析及typeof
chengxuyuancsdn
jstypeofjson解析
// json格式
var people='{"authors": [{"firstName": "AAA","lastName": "BBB"},'
+' {"firstName": "CCC&
- 流程系统设计的层次和目标
comsci
设计模式数据结构sql框架脚本
流程系统设计的层次和目标
- RMAN List和report 命令
daizj
oraclelistreportrman
LIST 命令
使用RMAN LIST 命令显示有关资料档案库中记录的备份集、代理副本和映像副本的
信息。使用此命令可列出:
• RMAN 资料档案库中状态不是AVAILABLE 的备份和副本
• 可用的且可以用于还原操作的数据文件备份和副本
• 备份集和副本,其中包含指定数据文件列表或指定表空间的备份
• 包含指定名称或范围的所有归档日志备份的备份集和副本
• 由标记、完成时间、可
- 二叉树:红黑树
dieslrae
二叉树
红黑树是一种自平衡的二叉树,它的查找,插入,删除操作时间复杂度皆为O(logN),不会出现普通二叉搜索树在最差情况时时间复杂度会变为O(N)的问题.
红黑树必须遵循红黑规则,规则如下
1、每个节点不是红就是黑。 2、根总是黑的 &
- C语言homework3,7个小题目的代码
dcj3sjt126com
c
1、打印100以内的所有奇数。
# include <stdio.h>
int main(void)
{
int i;
for (i=1; i<=100; i++)
{
if (i%2 != 0)
printf("%d ", i);
}
return 0;
}
2、从键盘上输入10个整数,
- 自定义按钮, 图片在上, 文字在下, 居中显示
dcj3sjt126com
自定义
#import <UIKit/UIKit.h>
@interface MyButton : UIButton
-(void)setFrame:(CGRect)frame ImageName:(NSString*)imageName Target:(id)target Action:(SEL)action Title:(NSString*)title Font:(CGFloa
- MySQL查询语句练习题,测试足够用了
flyvszhb
sqlmysql
http://blog.sina.com.cn/s/blog_767d65530101861c.html
1.创建student和score表
CREATE TABLE student (
id INT(10) NOT NULL UNIQUE PRIMARY KEY ,
name VARCHAR
- 转:MyBatis Generator 详解
happyqing
mybatis
MyBatis Generator 详解
http://blog.csdn.net/isea533/article/details/42102297
MyBatis Generator详解
http://git.oschina.net/free/Mybatis_Utils/blob/master/MybatisGeneator/MybatisGeneator.
- 让程序员少走弯路的14个忠告
jingjing0907
工作计划学习
无论是谁,在刚进入某个领域之时,有再大的雄心壮志也敌不过眼前的迷茫:不知道应该怎么做,不知道应该做什么。下面是一名软件开发人员所学到的经验,希望能对大家有所帮助
1.不要害怕在工作中学习。
只要有电脑,就可以通过电子阅读器阅读报纸和大多数书籍。如果你只是做好自己的本职工作以及分配的任务,那是学不到很多东西的。如果你盲目地要求更多的工作,也是不可能提升自己的。放
- nginx和NetScaler区别
流浪鱼
nginx
NetScaler是一个完整的包含操作系统和应用交付功能的产品,Nginx并不包含操作系统,在处理连接方面,需要依赖于操作系统,所以在并发连接数方面和防DoS攻击方面,Nginx不具备优势。
2.易用性方面差别也比较大。Nginx对管理员的水平要求比较高,参数比较多,不确定性给运营带来隐患。在NetScaler常见的配置如健康检查,HA等,在Nginx上的配置的实现相对复杂。
3.策略灵活度方
- 第11章 动画效果(下)
onestopweb
动画
index.html
<!DOCTYPE html PUBLIC "-//W3C//DTD XHTML 1.0 Transitional//EN" "http://www.w3.org/TR/xhtml1/DTD/xhtml1-transitional.dtd">
<html xmlns="http://www.w3.org/
- FAQ - SAP BW BO roadmap
blueoxygen
BOBW
http://www.sdn.sap.com/irj/boc/business-objects-for-sap-faq
Besides, I care that how to integrate tightly.
By the way, for BW consultants, please just focus on Query Designer which i
- 关于java堆内存溢出的几种情况
tomcat_oracle
javajvmjdkthread
【情况一】:
java.lang.OutOfMemoryError: Java heap space:这种是java堆内存不够,一个原因是真不够,另一个原因是程序中有死循环; 如果是java堆内存不够的话,可以通过调整JVM下面的配置来解决: <jvm-arg>-Xms3062m</jvm-arg> <jvm-arg>-Xmx
- Manifest.permission_group权限组
阿尔萨斯
Permission
结构
继承关系
public static final class Manifest.permission_group extends Object
java.lang.Object
android. Manifest.permission_group 常量
ACCOUNTS 直接通过统计管理器访问管理的统计
COST_MONEY可以用来让用户花钱但不需要通过与他们直接牵涉的权限
D