
管于TCGA数据库中的数据下载,我们之前有介绍过R语言下载包:R语言TCGA-Assembler包下载TCGA数据,同时在介绍数据库的使用教程中也介绍了在线下载以及官方下载工具下载:TCGA数据库使用教程。在线下载以及官方下载工具下载的数据是分开的,每个样本的数据的独立的,需要自己合并,这需要会R,Python 或者 perl 等编程语言(文末补充内容介绍)。
这里我们先介绍TCGAbiolinks包下载数据。因为这个包下载的数据是合并好的,不需要整理。
TCGAbiolinks下载TCGA数据
在第一讲我们介绍TCGAbiolinks包的时候,介绍了GDCquery这个函数,这是下载数据时要用到的函数,除此以外,我们还需要GDCdownload函数。GDCdownload函数使用GDC API或GDC传输工具下载GDC数据,用户可以使用查询参数查询的数据将保存在一个文件夹中:project/data.category。函数的整体框架为:
GDCdownload(query, token.file, method ="api", directory ="GDCdata",files.per.chunk =NULL)
各个参数介绍如下:
query:这个参数就是来自GDCquery的结果。
token.file:这个是下载受限的文件(仅适用于method=“client”),一般下载用不到。
method:使用API (POST方法)或gdc客户端工具。选择“api”,“client”。API更快,但是下载过程中数据可能会损坏,可能需要重新执行。
directory:下载数据的存放目录/文件夹。默认:GDCdata。
files.per.chunk:这将使API方法一次只下载n个(files.per.chunk)文件。当数据量过大时,可能会下载出错,可设置files.per.chunk参数减少下载问题。值为整数,即可将文件拆分为几个文件下载,如files.per.chunk = 6。
下面是一个下载数据的案例:
query <- GDCquery(project ="TCGA-ACC",data.category ="Copy number variation",legacy = TRUE,file.type ="hg19.seg",barcode = c("TCGA-OR-A5LR-01A-11D-A29H-01","TCGA-OR-A5LJ-10A-01D-A29K-01"))# 数据将被保存在 GDCdata/TCGA-ACC/legacy/Copy_number_variation/Copy_number_segmentationGDCdownload(query, method ="api")## Not run:# 从XML下载临床数据query <- GDCquery(project ="TCGA-COAD", data.category ="Clinical")GDCdownload(query, files.per.chunk = 200)query <- GDCquery(project ="TARGET-AML",data.category ="Transcriptome Profiling",data.type ="miRNA Expression Quantification",workflow.type ="BCGSC miRNA Profiling",barcode = c("TARGET-20-PARUDL-03A-01R","TARGET-20-PASRRB-03A-01R"))# 数据将被保存在:# example_data_dir/TARGET-AML/harmonized/Transcriptome_Profiling/miRNA_Expression_QuantificationGDCdownload(query, method ="client", directory ="example_data_dir")acc.gbm <- GDCquery(project = c("TCGA-ACC","TCGA-GBM"),data.category ="Transcriptome Profiling",data.type ="Gene Expression Quantification",workflow.type ="HTSeq - Counts"
总之,TCGAbiolinks包下载数据很简单,首先得明确自己要的是什么数据,通过GDCquery函数获取后,关于GDCquery请认真去学习上一讲:TCGA数据挖掘(一):TCGAbiolinks包介绍。对GDCquery了解后,再利用GDCdownload函数下载。这里说的相当简单,但聪明的人应该已经明白了。当然,我们后续的数据分析教程中还会更详细的介绍。
对于下载数据的分析可能会因自己的研究方向有所不同,有做甲基化的,有做SNP的等等,可以不用掌握全部,只需要会自己研究方向的即可,其他的做个了解,自己需要的时候,再学也不迟。
当然,我们后面会介绍一下常用的分析。
数据下载补充:数据整理
TCGAbiolinks包下载的数据是合并了的,不需要整理。在线下载或者官方工具下载的数据是分开的。我们介绍一下在线下载以及官方下载工具下载的数据怎么合并,这里用的是perl脚本,没有安装perl的可去官网:http://www.perl.org/get.html自行下载安装。这里你不需要懂perl语法,只需要知道DOS命令行的使用即可,脚本文末获取。
在线下载以及官方下载工具下载的数据是这样的。每一个文件夹是每个样本的数据,而且文件夹的名称和样本的barcode还不一致。

每一个文件夹里面的数据都是压缩包:

所以我们需要整理,要整理这些数据,首先我们在网页上筛选数据的时候,同时也要下载metadata这个文件。

下载metadata文件、数据文件以及脚本文件putFilesToOneDir.pl和mRNA_merge.pl在同一个文件夹。
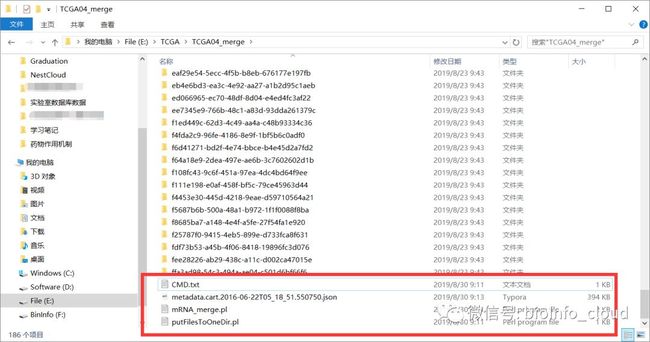
打开dos窗口,进入文件目录,键入:perl putFilesToOneDir.pl,回车。一会就会看见文件夹中多了一个files的文件夹,即我们将所有的数据都移动到了同一个文件夹下,当然,这个过程你可以手动。样本多的话,好像不科学。

进入files文件夹,里面全是压缩包,我们需要解压。这个就真的可以手动,因为每个人安装的压缩软件可能不一样,写脚本的话可能会报错,所以全选,解压到当前文件夹,也不是很费事。
下载metadata文件和perl脚本文件mRNA_merge.pl复制到files文件夹。

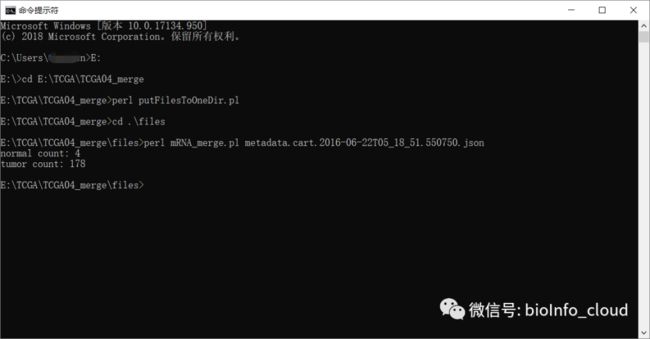
dos命令窗口下进入files文件夹,键入:perl mRNA_merge.pl metadata.cart.2016-06-22T05_18_51.550750.json,然后回车。等待时间与数据量有关。
运行结束后,在dos窗口会显示运行结果。normal count: 4;tumor count: 178,这里自己记下这2个参数,后面做分析的时候可能用到。
同时在files文件夹下生成了一个矩阵文件。这个文件我们后面做数据分析的时候可能会用到。

这里需要说明的是,这个脚本只适用于mRNA的Counts的数据。不适用于其他类型的数据。
扫码关注,后台回复:TCGA-mRNA_merge,领取案例文件和脚本文件。
