刘小泽写于19.1.16+19.1.19
随着不断地知识更新,之前不了解的东西开始变得熟悉,学习曲线就是这样
功能注释这么繁琐吗,有没有整合起来的流程?
之前看到,同源注释和功能分类都有各自的数据库和软件,并且同源注释还有两种方式,一步步操作会比较繁琐,因此就有软件看到了这个痛点,开发了新的整合式流程
-
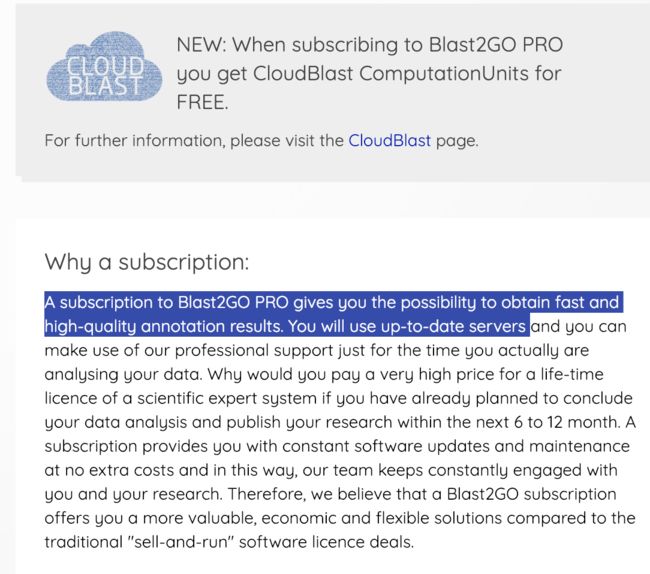
最有名的blast2go:https://www.blast2go.com/ 顾名思义,它是从blast直接给你GO分类结果,一步到位,但是目前专业版开始收费最便宜1600欧元/license,但提供一星期的试用。那么专业版好在哪里,官网也做了说明,主要还是强调:数据库更新快,分析结果更准确,还会送一个AWS云计算(https://www.blast2go.com/blast2go-pro/cloudblast 不需要用户自己安装数据库,及时更新,加速等等)
当然,基于blast软件,即使加持云计算优化,速度还是个问题;另外配置过程非常复杂
 blast2go
blast2go Trinotate:做过无参转录组的小伙伴都应该用过拼接转录本的Trinity吧,同样,这款软件也是Trinity团队为功能注释开发的(他们真的想从头到尾全实现了啊),这个软件的程序全是基于perl,所以不了解perl就不知道怎么解决报错
interpro+interproscan:部署也是个麻烦事,并且只支持基于结构域的注释,对KEGG、GO等数据库关联性不是很好
-
eggnog-mapper:2016年基因家族数据库EggNOG v4.5.1开始进入大家的视野,这个团队为数据库开发了相应的注释软件。目前已开发出5.0版本(http://eggnog5.embl.de/#/app/home) 支持了5,090个物种,比上一个版本4.5.1高出一倍不止。另外支持的Orthologous Groups也有4.5.1的19w疯狂升级到440W,其中每个Orthologous Group的蛋白也都关联了GO、KEGG数据库
 eggnog
eggnog
看来eggnog-mapper将是使用重点,那么它的设计原理是什么?
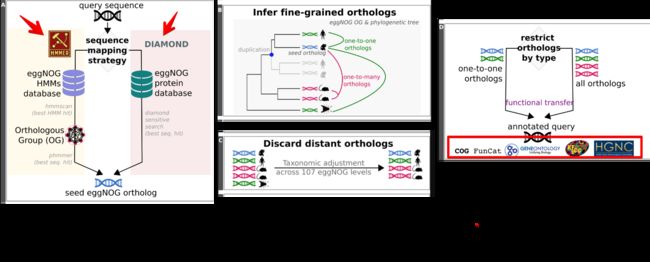
首先是比对,将要研究的序列与Eggnog中的蛋白序列进行比对,这个过程可以是基于相似性(DIAMOND),也可以是基于结构域(HMMER);
比对完以后就知道了我们提交的序列与哪个Orthologous Group最相似,并且还能知道与什么物种相似度最高,帮助确定注释的物种(真核还是原核,植物还是动物);
然后通过数据库之间关联进行COG、GO、Pathway的功能分类
安装eggnog-mapper
下载地址:https://github.com/jhcepas/eggnog-mapper/releases
另外还有详细的帮助文档:https://github.com/jhcepas/eggnog-mapper/wiki/Installation
或者直接conda安装eggnog-mapper

可以看到:这个软件基于python2.7,需要200G左右的硬盘空间,并且对于真核生物需要90GB的内存,细菌大约32G内存,古细菌10G内存 (因为使用快速模式的话,会把数据库一次性加到内存中)
下载数据库
它构建了107个各个层级的物种注释数据库
在这里http://eggnogdb.embl.de/#/app/downloads可以下载特有的数据库
当然也可以用download_eggnog_data.py euk bact arch viruses -y下载真核、细菌、古细菌、病毒(euk、bact、arch、viruses)的数据库
【如果是用conda安装的,命令可以直接使用;但自己手动安装的,需要把软件放入环境变量,并且还需要在开头加一个python2表示用python2进行编译】

下载完成看看,主要包括以下内容:
其中OG_fasta存储fasta文件;eggnog.db存储eggnog数据库中的蛋白序列ID与GO、KEGG等其他分类数据库的对应关系; eggnog_proteins.dmnd是diamond的数据库;hmmdb_levels这个文件夹存储HMMER的数据库
OG_fasta/ eggnog.db hmmdb_levels/
OG_fasta.tar.gz eggnog_proteins.dmnd og2level.tsv.gz
配置好了,然后看看怎么用
如果使用conda安装,直接激活安装的环境,然后emapper.py -h 查看帮助文档
-m {hmmer,diamond} Default:hmmer #设置模式,默认是hmmer
-i Input FASTA file containing query sequences #输入的文件FASTA
-d specify the target database #指定数据库(euk,bact,arch,viruses从中选择一个)
-o base name for output files #设置输出文件前缀
--usemem #[可选]针对hmmer模式(一次性将数据读入内存,快速处理,真核生物推荐90G以上内存服务器使用)
首先看比对模式有两种:diamond、hmmer
-
diamond(对于大量的序列比如多于1000条)
$ cat >diamond.sh emapper.py -m diamond \ -i test.fa \ -o diamond # 不需要指定数据库 -d -
hmmer(少量序列比对少于1000条)
$ cat >hmmer.sh emapper.py -m hmmer \ -i test.fa \ -d euk \ #(bact/arch/viruses) -o hmm-euk \ --usemem
实际运行下病毒的测试数据
这里选用的是Zaire ebolavirus病毒蛋白数据,因为很小所以比较好操作
数据下载
esearch -db protein -query PRJNA257197 | efetch -format=fasta > protein.fa
先构建小数据,随机选100条蛋白序列
$ seqtk sample protein.fa 100 > test.fa
# cat >hmmer.sh
$ emapper.py -m hmmer \
-i test.fa \
-d viruses \
-o hmm-vir \
--data_dir /YOUR_PATH/eggnog-mapper-1.0.3/data
# sh hmmer.sh 十几秒就运行完
# 然后把结果改一下,去掉#开头的行(但是保留query_name这一行的表头信息)
$ sed -i '/^# /d' hmm-vir.emapper.hmm_hits.annotations
# 然后再去掉query_name前面的#,并且去掉第一行的空行
$ sed -i 's/#//' hmm-vir.emapper.hmm_hits.annotations | sed '/^$/d'
得到了自己的eggnog-mapper结果后,只是搭建了一个桥梁,接下来就是构建自己的OrgDb,再进行GO、KEGG等富集分析
【在OrgDb这一步存在许多选择:
首先,如果研究的物种是模式物种,那么直接就可以用bioconductor上的注释包;
如果非模式物种而且有人构建过的话,就会上传到AnnotationHub,自己先用hub <- AnnotationHub()+display(hub)+query(hub, "拉丁名")看看上面有没有;
如果也没有,就可以利用eggnog的结果自己进行构建。当然即使AnnotationHub上有,我们也可以自行根据eggnog的结果进行构建,然后比较下和别人的结果】
欢迎关注我们的公众号~_~
我们是两个农转生信的小硕,打造生信星球,想让它成为一个不拽术语、通俗易懂的生信知识平台。需要帮助或提出意见请后台留言或发送邮件到[email protected]
