拿到测序数据后我们首先要进行质量评估(Quality Control),常用的工具就是FastQC。FastQC的详细使用说明:http://www.bioinformatics.babraham.ac.uk/projects/fastqc/Help/
该如何认识一个原始的测序数据(fastq data)呢?
一般我们可以从如下几个方面来分析:
- read各个位置的碱基质量值分布
- 碱基的总体质量值分布
- read各个位置上碱基分布比例,目的是为了分析碱基的分离程度
- GC含量分布
- read各位置的N含量
- read是否还包含测序的接头序列
- read重复率,这个是实验的扩增过程所引入的
FastQC支持的格式:
- FastQ (all quality encoding variants)
- Casava FastQ files
- Colorspace FastQ
- GZip compressed FastQ
- SAM
- BAM
- SAM/BAM Mapped only (normally used for colorspace data)
帮助文档:
# 基本格式# fastqc [-o output dir] [--(no)extract] [-f fastq|bam|sam] [-c contaminant file] seqfile1 .. seqfileN
# 主要是包括前面的各种选项和最后面的可以加入N个文件
# -o --outdir FastQC生成的报告文件的储存路径,生成的报告的文件名是根据输入来定的,注意是不能自动新建目录的。输出的结果是.zip文件,默认自动解压缩,命令里加上--noextract则不解压缩
# --extract 生成的报告默认会打包成1个压缩文件,使用这个参数是让程序不打包
# -t --threads 选择程序运行的线程数,每个线程会占用250MB内存,越多越快咯
# -c --contaminants 污染物选项,输入的是一个文件,格式是Name [Tab] Sequence,里面是可能的污染序列,如果有这个选项,FastQC会在计算时候评估污染的情况,并在统计的时候进行分析,一般用不到
# -a --adapters 也是输入一个文件,文件的格式Name [Tab] Sequence,储存的是测序的adpater序列信息,如果不输入,目前版本的FastQC就按照通用引物来评估序列时候有adapter的残留
# -q --quiet 安静运行模式,一般不选这个选项的时候,程序会实时报告运行的状况。
如果不加-q则会显示:
Started analysis of target.fq
Approx 5% complete for target.fq
Approx 10% complete for target.fq
......
如果输入的fastq文件名是test.fastq,fastqc的输出的压缩文件将是test.fastq_fastqc.zip。解压后,查看html格式的结果报告。

结果分析
查看html格式的结果报告。结果分为如下几项:

结果分为绿色的"PASS",黄色的"WARN"和红色的"FAIL"。
Basic Statistics

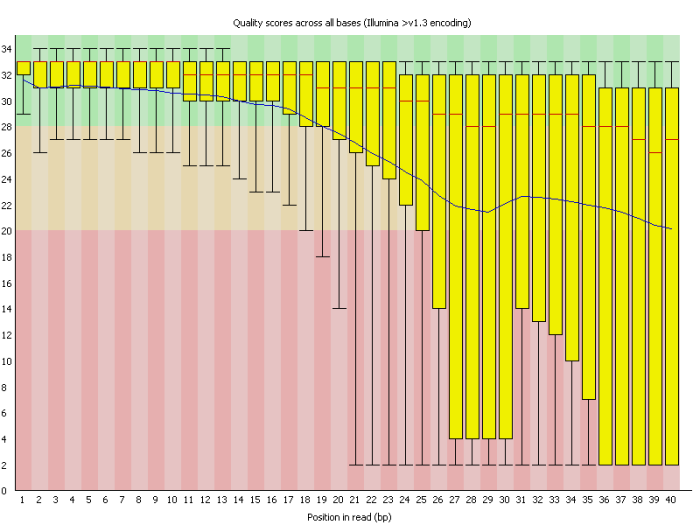
Per base sequence quality
横轴为read长度,纵轴为质量得分,Q = -10*log10(error P)。
Per tile sequence quality
每个tail测序情况,横轴表示碱基位置,纵轴表示tail的index编号,这个图主要是为了防止在测序过程中某些tail受到不可控因素的影响而出现测序质量偏低,蓝色表示测序质量很高,暖色表示测序质量不高。当某些tail出现暖色,在后续的分析种把该tail测序结果全部去除

Per sequence quality scores
横轴表示Q值,纵轴表示每个值对应的read数目,当测序结果主要集中在高分中,证明测序质量良好。

Per base sequence content
横轴为碱基长度分布,纵轴表示百分比,图中4条线分别代表A,C,T,G在每个位置上的平均含量。由于测序平台及测序长度不同,以及测序仪开始状态不稳定经常出现前后波动情况。

Per sequence GC content

红线是实际情况,蓝线是理论分布(正态分布,均值不一定在50%,而是由平均GC含量推断的),当红色出现双峰是表示混入了其他DNA序列。 曲线形状的偏差往往是由于文库的污染或是部分reads构成的子集有偏差(overrepresented reads)。形状接近正态但偏离理论分布的情况提示我们可能有系统偏差。
横轴表示GC含量,纵轴表示不同GC含量对应的read数
偏离理论分布的reads超过15%时,报"WARN";偏离理论分布的reads超过30%时,报"FAIL"。
Per base N content
当测序仪器不能辨别某条reads的某个位置到底是什么碱基时,就会产生“N”。对所有reads的每个位置,统计N的比率:

正常情况下N的比例是很小的,所以图上常常看到一条直线,但放大Y轴之后会发现还是有N的存在,这不算问题。当Y轴在0%-100%的范围内也能看到“鼓包”时,说明测序系统出了问题。当任意位置的N的比例超过5%,报"WARN";当任意位置的N的比例超过20%,报"FAIL"。
Sequence Length Distribution
reads长度的分布,当reads长度不一致时报"WARN";当有长度为0的read时报“FAIL”。

Sequence Duplication Levels
统计序列完全一样的reads的频率。测序深度越高,越容易产生一定程度的duplication,这是正常的现象,但如果duplication的程度很高,就提示我们可能有bias的存在(如建库过程中的PCR duplication)

横坐标是duplication的次数,纵坐标是duplicated reads的数目,以unique reads的总数作为100%。
当非unique的reads占总数的比例大于20%时,报"WARN";当非unique的reads占总数的比例大于50%时,报"FAIL“。
Overrepresented sequences
如果有某个序列大量出现,就叫做over-represented。fastqc的标准是占全部reads的0.1%以上。当发现超过总reads数0.1%的reads时报”WARN“,当发现超过总reads数1%的reads时报”FAIL“

Adapter Content
横轴表示碱基位置,纵轴表示百分比。当fastqc分析时没有选择参数-a adapter list时,默认使用图例中的4种通用adapter序列进行统计。若有adapter残留,后续必须去接头。


在构建测序文库时,会加上测序接头,其目的一方面是为了能够结合到flowcell上,另一方面是当有多个样本同时测序的时候能够利用接头信息进行区分。当测序read的长度大于被测序的DNA片段时,就会在read的末尾测到这些接头序列。一般的WGS测序是不会测到这些接头序列的,因为构建WGS测序的文库序列(插入片段)都比较长,约几百bp,而read的测序长度都在100bp-150bp这个范围。不过在进行一些RNA测序的时候,由于它们的序列本来就比较短,很多只有几十bp长(特别是miRNA),那么就很容易会出现read测通的现象,这个时候就会在read的末尾测到这些接头序列。
Kmer Content
重复短序列出现的次数

注意事项:
1、数据质控是一个综合的评价标准,其中主要指标为碱基质量与含量分布,如果这两个指标合格了,后面大部分指标都可以通过。如果这两项不合格,其余都会受到影响。
2、其中一些指标并不适合所有数据,例如DNA数据与RNA测序数据之间的差异等,要根据具体数据类型,具体分析。
以上就是一个完整的fastqC结果报告的简单说明
转载请注明出处
作者:oddxix
微信公众号:oddxix