加载之前下载并保存的文件
# 加载上一步从RTCGA.miRNASeq包里面提取miRNA表达矩阵和对应的样本临床信息。
load( file =
file.path(Rdata_dir,'TCGA-KIRC-miRNA-example.Rdata')
)
dim(expr)
dim(meta)
# 可以看到是 537个病人,但是有593个样本,每个样本有 552个miRNA信息。
# 当然,这个数据集可以下载原始测序数据进行重新比对,可以拿到更多的miRNA信息
# 这里需要解析TCGA数据库的ID规律,来判断样本归类问题。
group_list=ifelse(as.numeric(substr(colnames(expr),14,15)) < 10,'tumor','normal')
#substr(colnames(expr),14,15)提取列名字符串中14,15位字符(01代表肿瘤,11代表正常,约定好的)
#as.numeric将字符变为数字,一旦输出数字为01,小于10,则输出tumor,否则输出normal,从而得到和样本数一致的一列分组
table(group_list)
exprSet=na.omit(expr)#处理对象中缺失的值
source('../functions.R')
注意:这里介绍的差异分析方法有三种,其中limma是最经典的,但是limma是必须接受log之后的值,才能正确算出差异,一般芯片数据用limma包,(有些从GEO数据库下载的数据,经过标准化处理的时候是已经log过了的就不用log了)
测序的数据,DESeq2 ,edgeR一般用的比较多,接受的是RNAseq的count数据,非log的,如果数据已经log则要取log
方法一: Firstly run DESeq2
if(T){
library(DESeq2)
group_list<-as.factor(group_list)#把group_list变成因子,因为它接受的是因子
(colData <- data.frame(row.names=colnames(exprSet),
group_list=group_list) )#得到样本和分组对应的一个表
dds <- DESeqDataSetFromMatrix(countData = exprSet,
colData = colData,
design = ~ group_list)#构建模型
#tmp_f=file.path(Rdata_dir,'TCGA-KIRC-miRNA-DESeq2-dds.Rdata')
if(!file.exists(tmp_f)){
dds <- DESeq(dds)#差异分析前处理
save(dds,file ='DESeq2-dds.Rdata')#保存建模的结果
}
load(file = 'DESeq2-dds.Rdata') #直接加载建模结果

QQ截图20190714215742.jpg
res <- results(dds,
contrast=c("group_list","tumor","normal"))
#进行对比后提取数据,用results提取是因为看了说明书此函数的差异分析结果就是用这个提取
resOrdered <- res[order(res$padj),]#按照差异性大小排序
head(resOrdered)
DEG =as.data.frame(resOrdered)#变为数据框
DESeq2_DEG = na.omit(DEG) #删除NA值
nrDEG=DESeq2_DEG[,c(2,6)]#2为log2FoldChange 6为 pvalue 列,取出这两个
colnames(nrDEG)=c('log2FoldChange','pvalue')
draw_h_v(exprSet,nrDEG,'DEseq2',group_list,1)#draw_h_v是自定义的画图的函数,能画出火山图,PCI及热图
}
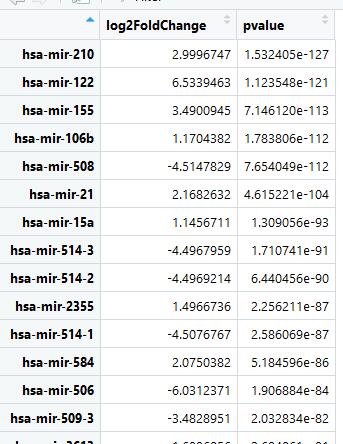
得到的差异分析结果
方法二:Then run edgeR (另一种差异分析方法)
if(T){
library(edgeR)
d <- DGEList(counts=exprSet,group=factor(group_list))#构建模型,只需要两个因素
keep <- rowSums(cpm(d)>1) >= 2#做筛选,此方法需要这样做
table(keep)
d <- d[keep, , keep.lib.sizes=FALSE]
d$samples$lib.size <- colSums(d$counts)
d <- calcNormFactors(d)
d$samples
dge=d
design <- model.matrix(~0+factor(group_list))
rownames(design)<-colnames(dge)
colnames(design)<-levels(factor(group_list))
dge=d
dge <- estimateGLMCommonDisp(dge,design)
dge <- estimateGLMTrendedDisp(dge, design)
dge <- estimateGLMTagwiseDisp(dge, design)
fit <- glmFit(dge, design)
# https://www.biostars.org/p/110861/
lrt <- glmLRT(fit, contrast=c(-1,1)) #因为是两组所以这么做,按照说明书
nrDEG=topTags(lrt, n=nrow(dge))
nrDEG=as.data.frame(nrDEG)
head(nrDEG)
edgeR_DEG =nrDEG
nrDEG=edgeR_DEG[,c(1,5)]
colnames(nrDEG)=c('log2FoldChange','pvalue')
draw_h_v(exprSet,nrDEG,'edgeR',group_list,1)
}
方法三: Lastly run voom from limma(第三种差异分析方法limma)
if(T){
suppressMessages(library(limma))
design <- model.matrix(~0+factor(group_list))
colnames(design)=levels(factor(group_list))
rownames(design)=colnames(exprSet)
design
dge <- DGEList(counts=exprSet)
dge <- calcNormFactors(dge)
logCPM <- cpm(dge, log=TRUE, prior.count=3)#注意limma包的表达数据是需要经过log的
v <- voom(dge,design,plot=TRUE, normalize="quantile")#说明书需要 normalize
fit <- lmFit(v, design)
group_list
cont.matrix=makeContrasts(contrasts=c('tumor-normal'),levels = design)#注意这里不要反了,反了就是相反的结果
fit2=contrasts.fit(fit,cont.matrix)
fit2=eBayes(fit2)
tempOutput = topTable(fit2, coef='tumor-normal', n=Inf)
DEG_limma_voom = na.omit(tempOutput)
head(DEG_limma_voom)
nrDEG=DEG_limma_voom[,c(1,4)]
colnames(nrDEG)=c('log2FoldChange','pvalue')
draw_h_v(exprSet,nrDEG,'limma',group_list,1)
}
#一路跑下来最终得到结果
tmp_f=file.path(Rdata_dir,'TCGA-KIRC-miRNA-DEG_results.Rdata')
if(file.exists(tmp_f)){
save(DEG_limma_voom,DESeq2_DEG,edgeR_DEG, file = tmp_f)
}else{
load(file = tmp_f)
}
nrDEG1=DEG_limma_voom[,c(1,4)]
colnames(nrDEG1)=c('log2FoldChange','pvalue')
nrDEG2=edgeR_DEG[,c(1,5)]
colnames(nrDEG2)=c('log2FoldChange','pvalue')
nrDEG3=DESeq2_DEG[,c(2,6)]
colnames(nrDEG3)=c('log2FoldChange','pvalue')
mi=unique(c(rownames(nrDEG1),rownames(nrDEG1),rownames(nrDEG1)))
lf=data.frame(lf1=nrDEG1[mi,1],
lf2=nrDEG2[mi,1],
lf3=nrDEG3[mi,1])
cor(na.omit(lf))
可以看到采取不同R包,会有不同的归一化算法,这样算到的logFC会稍微有差异。
算法不一样,可能得出不同的结果,通常把三个算法都跑一下,选交叉的基因
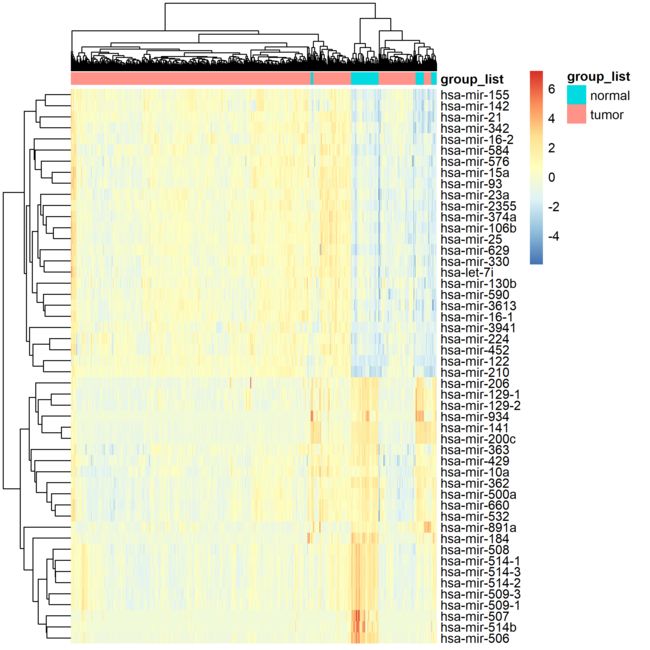
1_need_DEG_top50_heatmap.png
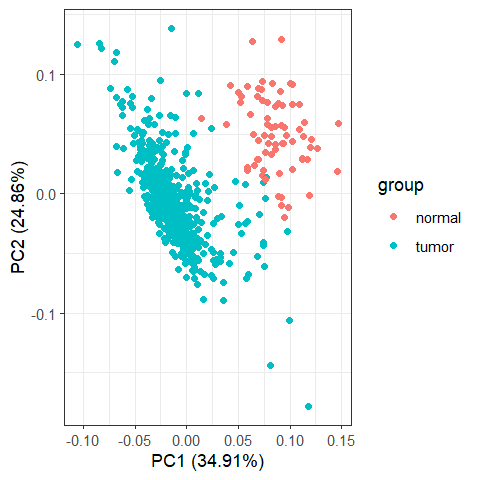
2_DEG_top50_pca.png
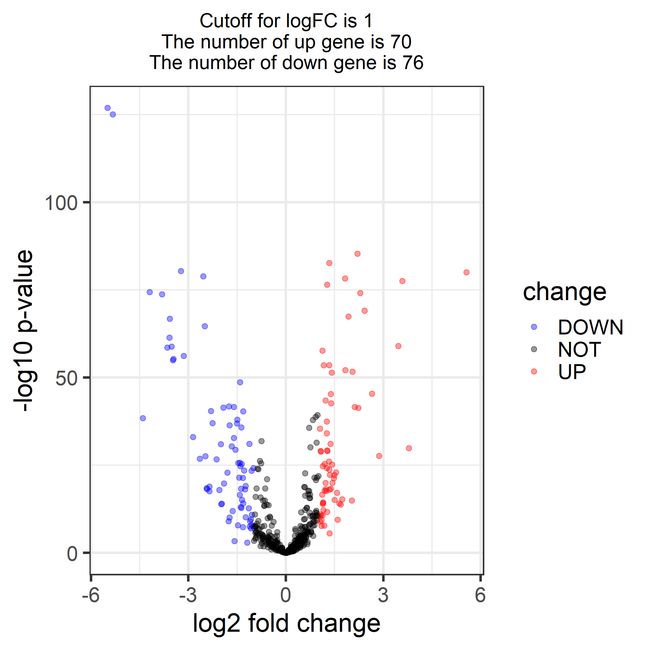
3_volcano.png
附 自定义函数draw_h_v代码:
draw_h_v <- function(exprSet,need_DEG,n='DEseq2',group_list,logFC_cutoff){
## we only need two columns of DEG, which are log2FoldChange and pvalue
## heatmap
need_DEG=nrDEG
library(pheatmap)
choose_gene=head(rownames(need_DEG),50) ## 50 maybe better
choose_matrix=exprSet[choose_gene,]#提取前50的表达矩阵
choose_matrix[1:4,1:4]
choose_matrix=t(scale(t(log2(choose_matrix+1)))) #画热图要先归一化
## http://www.bio-info-trainee.com/1980.html
annotation_col = data.frame( group_list=group_list )#变成一个表
rownames(annotation_col)=colnames(exprSet)#再取个名字
pheatmap(choose_matrix,show_colnames = F,annotation_col = annotation_col,
filename = paste0(1,'_need_DEG_top50_heatmap.png'))
library(ggfortify)
df=as.data.frame(t(choose_matrix))#行列转换
choose_matrix
df$group=group_list
png(paste0(2,'_DEG_top50_pca.png'),res=120)#存为什么文件
p=autoplot(prcomp( df[,1:(ncol(df)-1)] ), data=df,colour = "group")+theme_bw()#画图
print(p)
dev.off()
#画火山图
if(! logFC_cutoff){
logFC_cutoff <- with(need_DEG,mean(abs( log2FoldChange)) + 2*sd(abs( log2FoldChange)) )
}#如果没有给定阈值(差异倍数认为多少是上下调),则用平均值加上两倍的方差
logFC_cutoff=1
need_DEG$change = as.factor(ifelse(need_DEG$pvalue < 0.05 & abs(need_DEG$log2FoldChange) > logFC_cutoff,
ifelse(need_DEG$log2FoldChange > logFC_cutoff ,'UP','DOWN'),'NOT')
)
this_tile <- paste0('Cutoff for logFC is ',round(logFC_cutoff,3),
'\nThe number of up gene is ',nrow(need_DEG[need_DEG$change =='UP',]) ,
'\nThe number of down gene is ',nrow(need_DEG[need_DEG$change =='DOWN',])
)
library(ggplot2)
g = ggplot(data=need_DEG,
aes(x=log2FoldChange, y=-log10(pvalue),
color=change)) +
geom_point(alpha=0.4, size=1.75) +
theme_set(theme_set(theme_bw(base_size=20)))+
xlab("log2 fold change") + ylab("-log10 p-value") +
ggtitle( this_tile ) + theme(plot.title = element_text(size=15,hjust = 0.5))+
scale_colour_manual(values = c('blue','black','red')) ## corresponding to the levels(res$change)
print(g)
ggsave(g,filename = paste0(3,'_volcano.png'))
dev.off()
}