RNA_seq(1)植物转录组实战(下)之DESeq2进行差异基因分析
四、DESeq2差异基因分析
获得reads-counts之后,我们就可以开展差异基因分析了。我们以subread中的featureCounts工具得到的counts_id.txt为例,来进行后续的差异基因分析。
目前常见的差异基因分析工具有DESeq2、limma包等等,此处以DESeq2为工具来进行差异基因的筛选。
First step:获取表达矩阵和分组信息
进行差异基因分析之前,首先要获取表达矩阵和分组信息。我们的表达矩阵是刚才用featureCounts定量得到的counts_id.txt ,经过格式处理之后,是这样(部分截取):
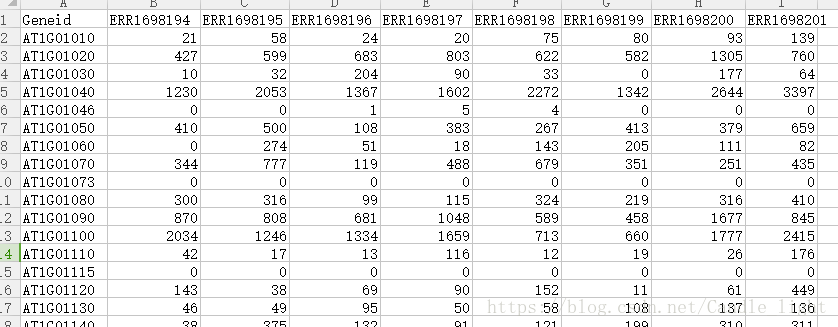
第一列是基因ID,之后的列都是样本ID
每一行代表不同的基因在不同样本中的表达量.
我们选day0和day1做比较,
为了方便,分组矩阵的制作我在R里面完成,输入如下代码:
us_count<-read.csv("C:\\Users\\admin\\Desktop\\RNA_seq_Ara_counts_day1_day0.csv",head=T,row.names=1) #输入表达矩阵数据路径
us_count<-round(us_count,digits=0) #将输入数据取整
#准备
us_count<-as.matrix(us_count) #将数据转换为矩阵格式
condition<-factor(c("day0","day0","day0","day0","day1","day1","day1","day1")) ## 设置分组信息,建立环境(8个样本,2组处理)
coldata<-data.frame(row.names=colnames(us_count),condition)
coldata #展示coldata内容
condition #展示环境上面的代码运行之后,我们的分组信息就是这样哒:

现在,就可以正式使用DESeq2做差异基因分析了,总共其实只有三步:
- 构建dds矩阵
- 对dds矩阵进行标准化
- 提取结果并绘制火山图
代码如下:
library(DESeq2) #使用library函数加载DEseq2包
##构建dds矩阵
dds<-DESeqDataSetFromMatrix(us_count,coldata,design=~condition)
head(dds) #查看构建好的矩阵
##进行差异分析
dds<-DESeq(dds) #对原始的dds进行标准化
resultsNames(dds) #查看结果名称
res<-results(dds) #用results函数提取结果,并赋值给res变量
summary(res) #查看结果
plotMA(res,ylim=c(-2,2))
mcols(res,use.names=TRUE)
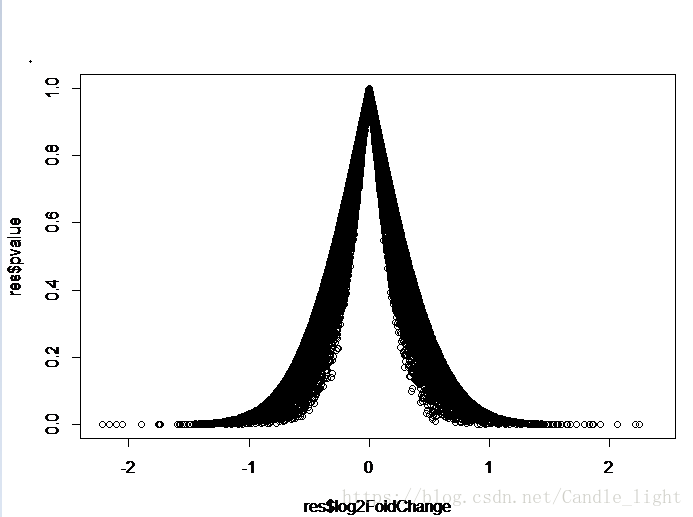
plot(res$log2FoldChange,res$pvalue) #绘制火山图
#提取差异基因
res <- res[order(res$padj),]
resdata <-merge(as.data.frame(res),as.data.frame(counts(dds,normalize=TRUE)),by="row.names",sort=FALSE)
deseq_res<-data.frame(resdata)
up_diff_result<-subset(deseq_res,padj < 0.05 & (log2FoldChange > 1)) #提取上调差异表达基因
down_diff_result<-subset(deseq_res,padj < 0.05 & (log2FoldChange < -1)) #提取下调差异表达基因
write.csv(up_diff_result,"C:\\Users\\admin\\Desktop\\上调_day0_VS_day1_diff_results.csv") #输出上调基因
write.csv(down_diff_result,"C:\\Users\\admin\\Desktop\\下调_day0_VS_day1_diff_results.csv") #输出下调基因至此,差异基因就成功提取了,看看火山图(火山图绘制是这句代码起效果:plot(res$log2FoldChange,res$pvalue) #其实就是对log2foldchange和pvalue作图):
五、 总结
自己之前处理线虫数据主要是用的RSEM(加bowtie2)来做RNA-seq的序列比对和生物学定量,但是没有接触过salmon和subread。这次使用这两个工具完成了植物的RNA-seq实战,对这些工具有了更深的理解,以后自己如果要开发相关软件,也要多用用,比较工具之间的优劣。