宏基因组测序流程(不完全版)
文章目录
- 所做工作
- 收获
- 宏基因组分析流程
- Step1.去除宿主污染
- Step2.去除接头序列
- Step3.对序列进行进一步质控
- Step4.对read进行进一步拼接(contig)
- Step5.对contig进行orf预测
- Step6.查找orf区翻译出来的氨基酸序列对应的蛋白质家族
所做工作
Step1.去除宿主污染
Step2.去除接头序列
Step3.对序列进⾏进⼀步质控
Step4.对read进⾏进⼀步拼接(contig)
Step5.对contig进⾏orf预测
Step6.查找orf区翻译出来的氨基酸序列对应的蛋⽩质家族
收获
- 基因组学基本知识
- 使用超算系统和在Linux下操作
- 下载和使用软件
- 理解各类常见数据格式的具体含义
- 学会使用在线网站
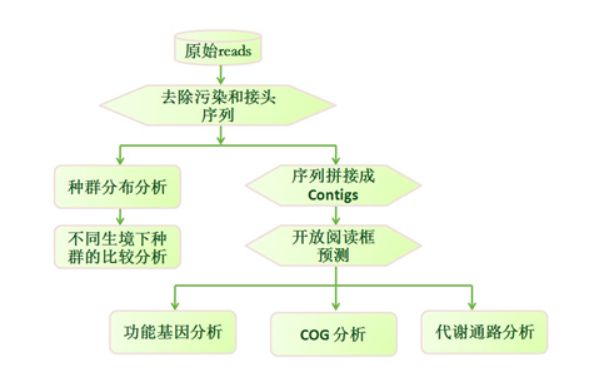
宏基因组分析流程
概述:宏基因组也称微生物环境基因组,定义为生境中全部微小生物遗传物质的总和。目前主要指环境样品中的细菌和真菌的基因组总和。宏基因组学就是一种以环境样品中的微生群物体基因组为研究对象, 以功能基因筛选和/或测序分析为研究手段, 以微生物多样性、 种群结构、 进化关系、 功能活性、 相互协作关系及与环境之间的关系为研究目的的新的微生物研究方法。
其具体流程大致如下图所示:
Step1.去除宿主污染
BWA是一款将DNA序列mapping到参考基因组上的软件,例如比对到人类基因组。其由三个算法组成:BWA-backtrack,BWA-SW和BWA-MEM。
使用BWA整个比对过程主要分为两步,第一步建立参考基因组也就是宿主基因组的索引,第二步使用BWA MEM进行比对。
-
BWA建索引
建索引有三种算法,通过参数-a is 、-a div和-a bwtsw进行选择。其中-a bwtsw对于短的参考序列是不工作的,必须要大于等于10Mb;-a is(效果和-a div是一样的)是默认参数,这个参数不适用于大的参考序列,必须要小于等于2G。
-
格式:
bwa index [-p prefix] [-a algoType] <in.db.fasta> -
实例:
# 第一步,下载hg19基因组作为参考基因组 wget http://hgdownload.cse.ucsc.edu/goldenPath/hg19/bigZips/chromFa.tar.gz tar zvfx chromFa.tar.gz cat *.fa > hg19.fa # 第二步,建立索引 ../tools/bwa-0.7.17/bwa index -a bwtsw -p /mnt/disk1/yang/yanglab/wangjn/data/index/hg19 hg19.fa
-

-
使用BWA MEM进行比对
-
格式:
bwa mem [-aCHMpP] [-t nThreads] [-k minSeedLen] [-w bandWidth] [-d zDropoff] [-r seedSplitRatio] [-c maxOcc] [-A matchScore] [-B mmPenalty] [-O gapOpenPen] [-E gapExtPen] [-L clipPen] [-U unpairPen] [-R RGline] [-v verboseLevel] db.prefix reads.fq [mates.fq]- 命令:
../../tools/bwa-0.7.17/bwa mem -t 16 -M /mnt/disk1/yang/yanglab/wangjn/data/index/hg19 liweiling2016-10-25-shu-qian-2.read1_Clean.fastq.gz liweiling2016-10-25-shu-qian-2.read2_Clean.fastq.gz 1>liweiling2016-10-25-shu-qian-2.sam
-

- 查看sam文件中和人类基因组比中的序列。
J00103:120:HFFJLBBXX:8:1101:6340:1613 163 chr11 55386856 0 34S19M97S = 55386856 19 ATAAATCCGTTGCATGTACATTTTTGCTGTTATGGGTGTTCGTTTTTTTGTCCGCTGACTTGTTTGCCGGTTCATTCCGCACCTTGGGTATCAAGGAGGGATTGGGCAGTCGGCAAGTATTCCAGATAAACAAGGATTCAGCCGGATTTG AAFFFJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJFJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJFJJJJJJJJJJJJJJFJJJJJJJJJJJJJJ<JJJJJFFJFJJJJJJJJJJFJJJJJJJJFF7FJJJJJJJJJJJJJJFFJJJ NM:i:0 MD:Z:19 MC:Z:28S19M103S AS:i:19 XS:i:0
比如说对于上面的这条结果,就是名称为J00103:120:HFFJLBBXX:8:1101:6340:1613的read与11号染色体的55386856位置比中。
-
使用samtools去除宿主序列
-
将sam文件转换为bam文件
-
参数说明
-b:以bam格式输出
-u:以未压缩的bam格式输出,一般与linux命令配合使用
-h:在sam输出中包含header信息
-H:只输出header信息
-f [INT]:只输出在比对flag中包含该整数的序列信息
-F [INT]:跳过比对flag中含有该整数的序列
-o [file]:标准输出结果文件
-S : 默认下输入是 BAM 文件,若是输入是 SAM 文件,则最好加该参数,否则有时候会报错
-
命令
samtools view -bS liweiling2016-10-25-shu-qian-2.sam > liweiling2016-10-25-shu-qian-2.bam
-
-
筛选出没有比中的序列
-
bam的flag图示,上面的标签都是2的n次方,这样的数有一个特点,就是随机挑选其中的几个,它们的和是唯一的。比如要筛选未比中的序列,由于4和8都代表未比中,因此筛选目标为4+8=12。
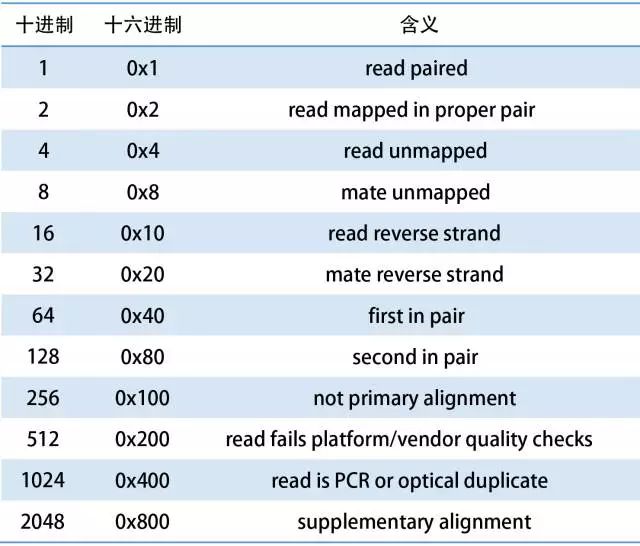
1 : 代表这个序列采用的是PE双端测序
2: 代表这个序列和参考序列完全匹配,没有插入缺失
4: 代表这个序列没有mapping到参考序列上
8: 代表这个序列的另一端序列没有比对到参考序列上,比如这条序列是R1,它对应的R2端序列没有比对到参考序列上
16:代表这个序列比对到参考序列的负链上
32 :代表这个序列对应的另一端序列比对到参考序列的负链上
64 : 代表这个序列是R1端序列, read1;
128 : 代表这个序列是R2端序列,read2;
256: 代表这个序列不是主要的比对,一条序列可能比对到参考序列的多个位置,只有一个是首要的比对位置,其他都是次要的
512: 代表这个序列在QC时失败了,被过滤不掉了(# 这个标签不常用)
1024: 代表这个序列是PCR重复序列(#这个标签不常用)
2048: 代表这个序列是补充的比对(#这个标签具体什么意思,没搞清楚,但是不常用)
- 命令
samtools view -b -f 12 -F 256 liweiling2016-10-25-shu-qian-2.bam > liweiling2016-10-25-shu-qian-2-Unmapped.bam- 含义
筛选出两端都没有比对上的序列,遇到不是主要比对的序列直接跳过。
-
-
对bam进行排序
-
说明
samtools sort -n -m
-m 参数默认下是 500,000,000 即500M(不支持K,M,G等缩写)。处理大数据时,如果内存够用,则设置大点的值,以节约时间。-n 设定排序方式按short reads的ID排序。默认下是按序列在fasta文件中的顺序(即header)和序列从左往右的位点排序
-
使用
samtools sort -n liweiling2016-10-25-shu-qian-2-Unmapped.bam -o liweiling2016-10-25-shu-qian-2-Unmapped_sorted.bam
-
-
使用bedtools将bam转换成fastq
bedtools bamtofastq -i liweiling2016-10-25-shu-qian-2-Unmapped_sorted.bam -fq liweiling2016-10-25-shu-qian-2_removed_r1.fastq -fq2 liweiling2016-10-25-shu-qian-2_removed_r2.fastq -
Step2.去除接头序列
可以采用SeqPrep程序合并重叠成单个较长读数的成对末端Illumina读数。也可以仅对adapter进行修剪,而不进行任何配对的末端重叠。
- 必要的输入参数:
- -f first read input fastqfilename #正链输入文件
- -r second read input fastq filename #反链输入文件
- -1 first read output fastq filename #正链输出文件
- -2 second read output fastq filename #反链输出文件
- 可选的输入参数:
- -3 first read discarded fastq filename #正链丢弃的文件
- -4 second read discarded fastq filename #反链丢弃的文件
- -6#输入序列的格式,默认为phred+33,设置后为phred+64
- -q Quality score cutoff for mismatches to be counted in overlap;default = 13 #质量分数
- -L Minimum length of a trimmed or merged read to print it; default= 30 #最小长度
- -O,-M,-N,-Q…
- 关于adapter的控制参数:
- -A #正链序列的adapter,默认值为AGATCGGAAGAGCGGTTCAG
- -B #反链序列的adapter,默认值为AGATCGGAAGAGCGTCGTGT
- 关于输出结果的控制参数:
- -y #最大化质量分数
- -s #指定将merge后的结果合并到哪个文件
- -E,-x, -o, -m, -n指的是对输出结果进行进一步的细化操纵
../../tools/SeqPrep/SeqPrep -f liweiling2016-10-25-shu-qian-2_removed_r1.fastq -r liweiling2016-10-25-shu-qian-2_removed_r2.fastq -1 liweiling2016-10-25-shu-qian-2_removed_noadapter_r1.fq.gz -2 liweiling2016-10-25-shu-qian-2_removed_noadapter_r2.fq.gz -s liweiling2016-10-25-shu-qian-2_removed_noadapter.fq.gz
# 解压文件
gunzip liweiling2016-10-25-shu-qian-2_removed_noadapter.fq.gz
mv liweiling2016-10-25-shu-qian-2_removed_noadapter.fq liweiling2016-10-25-shu-qian-2_removed_noadapter.fastq
Step3.对序列进行进一步质控
FASTX-Toolkit是用于短读FASTA / FASTQ文件预处理的命令行工具的集合。FASTQ Information可用来执行图表质量统计和核苷酸分布,FASTQ/A Collapser可用来将FASTQ / A文件中的相同序列折叠成单个序列(同时保持读取计数)。FASTQ/A Clipper用来删除测序适配器/连接器。FASTQ Quality Filter用来根据质量过滤序列。要注意的一点是:不支持压缩格式的输入文件,不允许序列中存在N碱基,这样的序列会自动去除
- fastx_clipper去除adapter
- fastx_collapser比对基因组,去除重复序列
- fastx_quality_stats对质量值进行统计
- fastq_quality_boxplot_graph.sh绘制碱基质量分布盒式图
- fastx_nucleotide_distribution_graph.sh绘制碱基分布图
fastx_collapser -v -i liweiling2016-10-25-shu-qian-2_removed_noadapter.fastq -o liweiling2016-10-25-shu-qian-2_removed_noadapter_collapsed.fastq && fastx_quality_stats -i liweiling2016-10-25-shu-qian-2_removed_noadapter_collapsed.fastq -o liweiling2016-10-25-shu-qian-2_removed_noadapter_collapsed.txt && fastq_quality_boxplot_graph.sh -i liweiling2016-10-25-shu-qian-2_removed_noadapter_collapsed.txt -o lliweiling2016-10-25-shu-qian-2_removed_noadapter_collapsed_quality.png -t "liweiling2016-10-25-shu-qian-2_removed_noadapter_collapsed" && fastx_nucleotide_distribution_graph.sh -i liweiling2016-10-25-shu-qian-2_removed_noadapter_collapsed.txt -o liweiling2016-10-25-shu-qian-2_removed_noadapter_collapsed_nuc.png -t "liweiling2016-10-25-shu-qian-2_removed_noadapter_collapsed"
查看图片
eog lliweiling2016-10-25-shu-qian-2_removed_noadapter_collapsed_quality.png
eog liweiling2016-10-25-shu-qian-2_removed_noadapter_collapsed_nuc.png


Step4.对read进行进一步拼接(contig)
细菌基因组组装工具之SPAdes SPAdes是由俄罗斯科学院圣彼得堡理工大学计算生物学实验室开发的基因组拼接工具。主要用于组装细菌基因组、二代(宏)基因组、(宏)转录组测序的拼接,也可用于一、二、三代测序的混合组装。是目前评价最好的拼接工具之一。
参数说明
-
Basic options:
-
-o 输出文件夹
-
–sc this flag is required for MDA (single-cell) data
-
–meta 这个是用于组装宏基因组**
-
–rna this flag is required for RNA-Seq data
-
–plasmid runs plasmidSPAdes pipeline for plasmid detection
-
–iontorrent this flag is required for IonTorrent data
-
–test runs SPAdes on toy dataset
-
-h/–help prints this usage message
-
-v/–version prints version
-
-
Input data:
-
–12 正向和反向合在一起的paired-end reads
-
-1 paired-end reads的正向序列
-
-2 paired-end reads的反向序列
-
-s unpaired reads的序列
-
-
Pipeline options:
-
–only-error-correction runs only read error correction (without assembling)
-
–only-assembler runs only assembling (without read error correction)
-
–careful tries to reduce number of mismatches and short indels
-
–continue continue run from the last available check-point
-
–restart-from restart run with updated options and from the specified check-point (‘ec’, ‘as’, ‘k’, ‘mc’)
-
–disable-gzip-output forces error correction not to compress the corrected reads
-
–disable-rr disables repeat resolution stage of assembling
-
-
Advanced options:
- –dataset file with dataset description in YAML format
- -t/–threads 线程数,默认是16个
- -m/–memory RAM limit for SPAdes in Gb (terminates if exceeded) [default: 250]
- –tmp-dir directory for temporary files [default: /tmp]
- -k k-mer的设置数,如果是多个,中间要以逗号隔开,必须为奇数且不能大于128,即最大设置数 为127 [default: ‘auto’]
- –cov-cutoff coverage cutoff value (a positive float number, or ‘auto’, or ‘off’) [default: ‘off’]
命令
/mnt/disk1/yang/yanglab/wangjn/tools/SPAdes-3.13.0-Linux/bin/spades.py -1 liweiling2016-10-25-shu-qian-2_removed_noadapter_r1.fq.gz -2 liweiling2016-10-25-shu-qian-2_removed_noadapter_r2.fq.gz -t 20 -o liweiling2016-10-25-shu-qian-2_removed_noadapter
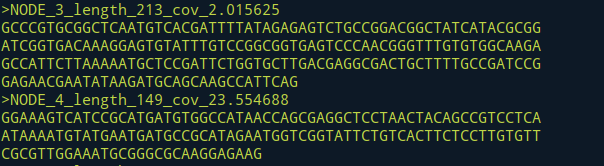
Step5.对contig进行orf预测
下载了ORFfinder之后发现服务器下不能直接运行可执行文件,所以以线上预测代替。
网址:https://www.ncbi.nlm.nih.gov/orffinder/
- 以Kmer值为21时计算得来的contig结果进行预测为例
原始序列
>NODE_6_length_670_cov_2.385208
TCCCACAAGATTGATATAATAATTTTCCAGACTTTCATCACATTCCTGCATAGCTTTAAT
CTTGCAATTTTTCTTGTCAAGAGTCAGCGCCAAATCTGTTACAGAAATACTGCTGAAAAT
ATCAGCCTCTGTTTGAGATAAAACAGTATACTCTACTCCCATTTCATCTAATACAAAGCT
CAAAGTTTTCGTGTCAGACACTTCTATGTGGATACATTTACGGCAGGCTTTTTCCAATTC
AACTGCACTGATTTCCTTTACAATACATCCTTTATCAATAAATCCAAAATGTGTTGCAAT
TTTTGACAACTCATCTAAAATGTGACTGGAAATCAAAACAGTAATCCCCTGCTCCCGATT
AAGCTTTAAAATCAATTCTCGAATTTCAATCATTCCTTGAGGGTCTAATCCATTTACCGG
TTCGTCTAAAATCAAAAAGTCCGGATTTCCTGCAAGTGCAACGGCAATTCCCAGTCTCTG
CCGCATACCCAGAGAAAAATTTTTTGCCTTTTTCTTACCGGTATTTTCCAGTCCCACAAG
CTTCAAAATATCAGTAATTCCTTCAAAAGACGGCATTCCTAGATACCGATATTGTGCTTT
CATATTATCTTCTGCCGTCATATCCAGATAAATAGCCGGTGTTTCCACCACAGCCCCTAT
TCTTTTCCTT
计算结果

Step6.查找orf区翻译出来的氨基酸序列对应的蛋白质家族
网址:http://www.ncbi.nlm.nih.gov/Structure/cdd/wrpsb.cgi
上文计算出来的ORF2对应翻译的氨基酸序列
>lcl|ORF2
MTAEDNMKAQYRYLGMPSFEGITDILKLVGLENTGKKKAKNFSLGMRQRL
GIAVALAGNPDFLILDEPVNGLDPQGMIEIRELILKLNREQGITVLISSH
ILDELSKIATHFGFIDKGCIVKEISAVELEKACRKCIHIEVSDTKTLSFV
LDEMGVEYTVLSQTEADIFSSISVTDLALTLDKKNCKIKAMQECDESLEN
YYINLVG
查找结果
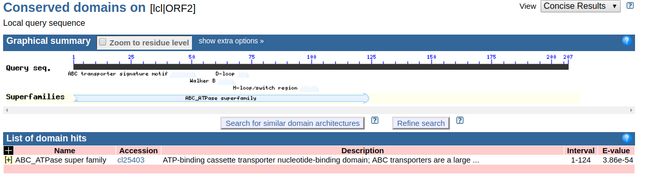
可以看到该段氨基酸序列由较大概率隶属于ABC_ATPase家族