转录组分析的正确姿势
(蓝色文字都可点击跳转到对应推文)
转录组分析是目前应用最广的高通量测序分析技术之一。常见设计是不同样品之间比较,寻找差异基因、标志基因、协同变化基因、差异剪接和新转录本,并进行结果可视化、功能注释和网络分析等。
转录组的测序分析也相对成熟,从RNA提取、构建文库、上机测序再到结果解析既可以自己完成,又可以在专业公司进行。
概括来看转录组的分析流程比较简单,序列比对-转录本拼接 (可选)-表达定量-差异基因-功能富集-定制分析。整个环节清晰流畅,可以作为最开始接触高通量测序学习最合适的技术之一。
但重点和难点在于理解这些过程都是怎么做的,有什么需要注意的,结果怎么解读,后续分析怎么做。这些只有自己动手操作过,才可能有理解。而理解了一个,再去做其它类型分析,也会轻松很多。
而且现在三代测序火起来了,该怎么去选择呢? 三代测序能帮我们解决什么问题,不能做什么,有什么需要注意的,分析起来有什么不同,二代-三代如何统一分析?也是我们面临的一个新问题。
实验设计这块重要的是对照和至少3个生物学重复,并选择合适的测序通量。ENCODE要求重复之间的Spearman correlation值大于0.9 (遗传背景不一致的生物重复相关系数要大于0.8)。定量基因表达和评估转录图谱相似性只需要中等测序深度;而研究新转录本和可变剪接则需要更深的测序;一般来讲长RNA-seq文库测序深度满足可用reads在20-30 million (如果测PE150,换算成碱基数为6G-9G)。
另外一个需要注意的是测序的批次效应,保证自己的样品同时处理、RNA同时提取、同时构建文库和上机测序。这些环节虽然不能总受我们控制,但记录下对应的操作时间和批次,最后在绘制表达图谱时与实验相关参数进行关联展示 (利用我们介绍的热图简化或高颜值可定制在线绘图工具-第三版),从而保证结果没有受到试验中处理批次的影响。ENCODE计划有一篇文章在比较人和小鼠不同组织的表达谱相似度时得到的结果是样品按物种而非组织聚在一起,这与之前认为的发育通路的保守性不符。后来发现是测序批次捣的鬼,做了批次效应矫正后,表达图谱按组织而非物种聚在一起了。
测序环节通常不需要自己操作,测序公司都很成熟,但测序的原理需要知道。这会影响到后续分析时参数的选择,比如知道什么是插入片段大小,什么是链特异性测序,什么情况会有接头序列,双端测序如何测等。
获得数据后,就涉及到数据的传输和质量评估(也包括如何从公共数据库下载数据)和文件格式的转换。FASTQ格式解释和质量评估中有些提及。质量评估的意义在于从测序质量角度评价建库和测序的成功与否,指导接头和低质量碱基的去除。这一步参数控制的严格与否对后续的比对会有影响,同时也会受到后续分析选择的工具的影响。对Linux系统一定程度的了解,是进行这些工作的基础。
39个转录组分析工具,120种组合评估(转录组分析工具哪家强)中讲述了如何选择、评估合适的比对工具,序列拼装工具,定量工具和差异分析工具。值得我们在进入正式的分析之前,仔细阅读。另外类似的评估文章,还有几篇,都可以一并读一下,这样在后期分析时对工具的选择和使用才更得心应手。
工具比较类文章一般只告诉你做了什么,不告诉你这么做的原因是什么,而且每一步细分开来又有很多小细节需要注意,比如在比对环节就会涉及到:不同的样本如何选择合适的基因组和注释文件,什么样的软件支持Junction reads的比对,什么样的比对率是合适的,比对质量怎样,测序中RNA有无降解或选择偏好性,测序饱和度如何等。
这些可能都不会体现在最终的结果中,但都是确保后期结果可靠性所必须要做的事情。2002年诺贝尔奖得主Sydney Brenner曾对数据分析做过提醒Garbage in, Garbage out。软件是死的,提供了格式正确的输入,就可以得到输出,但输出正确与否,就得靠人的经验来判断了。
在后面的差异基因鉴定阶段,还存在把FPKM值转换为整数再提交给DESeq2做分析的,软件不报错,但结果不对。或者能顺着教程运行DEseq2分析,但换成自己的数据就不知道如何下手的。这些问题都需要在实践过程中持续不断的试错、阅读更多的文章和教程来步步矫正。这当然是一个耗时耗力的过程,那么有没有一个更好的方式呢?
生信宝典团队经过紧张的筹备,决定推出一系列的针对生信学习和高通量分析的兴趣小组(在生信学习系列教程的基础上进一步拓展和深入),跟大家一起去走过这段历程。我们的口号是易生信,毕生缘,希望能通过短暂高强度的训练快速推进大家在生信分析领域的进展。
但生信学习是个缓慢的过程,需要教、学、练、改不断的循环。我们希望能通过系列课程,再加上四段式培训模式集中讲解实战(2天)-自行练习(5天)-再讲解答疑考核(2天)-后续视频观摩和群内讨论跟大家一起探索如何尽可能快的学会生信,学到可以自己做,有问题自己可以解决的程度。点击阅读原文可查看详细信息。
课程简介
一、转录组的应用、设计和案例分享
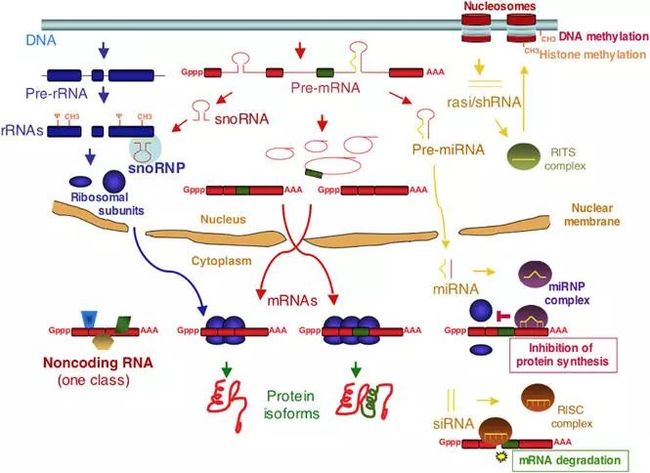
转录组学研究技术介绍
转录组学实验设计和测序原则、注意事项
转录组学文章案例分析
在线基因表达资源数据库
二、转录组分析流程实战
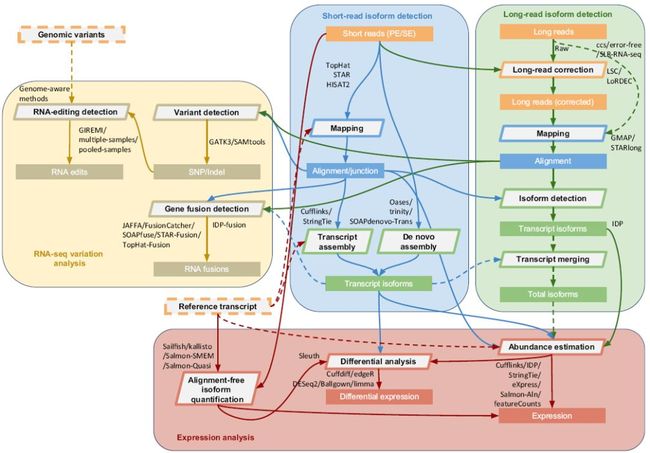
测序数据质量评估和清洗
基于比对的差异基因分析
不基于比对的差异基因分析
转录本组装和选择性剪接分析
目标基因富集分析
三、转录组高级分析

WGCNA基因共表达分析
WGCNA基因、表型关联分析
Cytoscape 共表达网络绘制
转录组常见图形在线绘制
四、三代测序技术概述

PacBio和Oxford Nanopore测序的原理
三代测序的特点和应用
三代测序在转录组研究的优势和案例分享
五、三代测序基本分析流程

原始测序序列去除接头和错误序列
提取环形一致序列读长(CCS reads)
CCS reads分类(包括全长和非全长CCS reads)
CCS reads聚类(根据CCS reads序列的相似性)获得最终的转录本集合
最终转录本比对回基因组
转录本定量和可变剪接分析
如果您有其它关注的问题,也请报名时提出,把这次课程变成您的定制讲解。
主讲教师
主讲老师包括爱荷华大学、中科院微生物所、遗传发育所、基因组所、生物物理所等多名本领域一线技术专家。
助教团队
十余名科学院、清华、北大博士(含在读),轮值讲师和助教,辅助学员学习和矫正培训过程中不足的点。
培训时间
2018-05-12 到 2018-05-13 (主要是二代测序)
2018-05-19 到 2018-05-20 (主要是三代和二三混合)
独创线下集中授课2天+自行练习5天+再集中讲解答疑2天+后期学习群的四段式教学,并提供学习视频,教、学、练、答结合,真正实现独立分析大数据。
每天早9点到晚5点,半封闭式教学
报到时间:上课当天。
授课地点
北京市西城区鼓楼明德大厦 (北京市旧鼓楼大街47号院2号楼2010)。
课程价格
限时优惠4199元/人
之后恢复原价6999元/人 (住宿自行解决,提供培训期间午餐)名额有限,每次课程报名满30人后自动关闭报名通道
提供易汉博基因科技实习机会或工作机会
促销优惠活动
座位按报名并成功缴费顺序从前到后龙摆尾式排序
赠送价值188元线上生信基础课程一门,目前的《应用Python处理生物信息数据和作图》、《生物信息作图系列R、Cytoscape及图形排版》和《生物信息中的Linux应用》任选其一。(http://bioinfo.ke.qq.com)
多人(N,10>N>1)组团报名并同时缴费,每人还可获得价值N百元(最高500)的礼品(充值或购物卡)。
更多课程的详细介绍,请扫描下方二维码。

复制以下链接
http://www.ehbio.com/Training/ 或
点击阅读原文跳转报名页