扩增子16S分析专题研讨论会——背景介绍
整理一下我近期报告的PPT、文稿和视频,分享给大家,希望对同行有所帮助。
本节课程视频共分3部分。
https://v.qq.com/x/page/t3015tp7d5u.html
Part 1. 21扩增子分析背景介绍p1-23,23min
https://v.qq.com/x/page/j3015gkf92g.html
Part 2. 21扩增子分析背景介绍p24-39,32min
https://v.qq.com/x/page/m3015en6o80.html
Part 3. 21扩增子分析背景介绍p40-56.wmv,25min

大家好,我叫刘永鑫,来自中国科学院遗传与发育生物学研究所,今天很高兴有这次机会为大家来讲扩增子分析系列课程。我本科学习的是微生物学专业,之后又获得了生物信息学博士学位,在短暂的两年博士后科研工作后,留所任工程师,主要负责宏基因组学的数据分析。在过去的两年工作里,主要参与并发表的文章有10余篇,累积影响因子150多分,其中包括一篇Science和两篇Nature Biotechnology。同时还是宏基因组公众号的创始人,在两年多的时间里,分享了400多篇原创文章,写作量超过200万字,阅读量超过1000多万次。我们接下来让大家一次对自己的研究方向,姓名和单位进行简单自我介绍,方便大家的沟通。
很感谢大家对自己基本情况和研究方向的介绍,这对于我下面课程中和重点的突出很在帮助,也希望同行互相认识,多交流和互相帮助。下面我们开始今天的课程,本次为第2天的第1节课,主要介绍扩增子分析的背景知识,右边这个图是来自2016年一篇Nature Protocol的文章,对微生物组近10年的发展进行了总结,我们可以看到从2010年到2016年我们开始对哪些环境对象进行探索,包括极端环境、植物叶片、白蚁、人类肠道、海洋、永久冻土、以及土壤沉积物的研究,这个领域扩展到了我们所能探索的所有地方。

先给大家看一幅漫画,这张图最能代表我这10年对科研的感悟,我在本科的时候,学了4年的微生物学,之后又花了6年,获得了生物学博士学位,然后做了两年博士后科研工作,用了12年的时间,我以为我走到了一个科研的很专业的高度。其实只是给我看到微生物组这个领域的入场券,在正式工作后加入了微生物组这个领域,我发现自己有多渺小,之前学的东西是那么的微不足道,微生物组学是一个非常伟大的领域,有永远挖不完的宝藏。在这个领域里工作的越久越会发现他有多么深不可测,而且你越会发现自己有太多的东西不知道。

本节课主要从以下4个方面进行介绍,包括为什么要学习生物信息,什么是微生物组?扩增子测序的基本概念以及扩增子分析的基本思路。

首先介绍,我们为什么要学习生物信息学
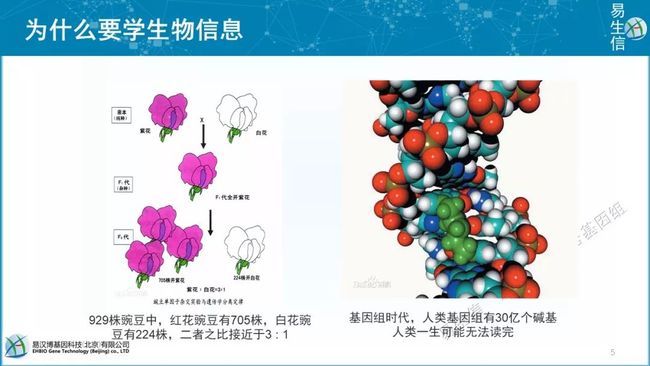
我们为什么要学习生物新学呢?举一个简单的例子,我们在高中都学过孟德尔发现了遗传学的基本定律及基因分离定律和基因自由组合定律,他研究的对象就是豌豆,他主要靠紫花的豌豆和白花的豌豆进行杂交,再自交,获得的子代有1000多株进行简单的颜色数量统计,发现了3:1的规律,最终确定了基因的分离定律。而在基因组学时代,我们人类基因组的单倍体就有30亿个碱基对,如果把它当成一本书的话我们可能一生都无法读完。

如果你觉得人类基因组有30个碱基对已经是天文数字的话。但本质上我们人类的基因组只有25,000多个编码基因,而我们的肠道微生物组,却有多达1000万个编码基因,典型的有300~500种微生物。各种海量的微生物组数据,我们必须借助计算机每秒上亿次的计算速度,甚至是超级计算机来帮我们完成这种大规模数据的分析。

我们要使用计算机,就要对计算机的基本结构有一定的了解,计算机最主要的三个核心硬件就是CPU、内存和硬盘,CPU的单位本来是GHz,但现在的单核计算速度已经发展到了瓶颈,主要的参数是CPU核心数量或线程数。你应该经常听到笔记本、手机四核、8核,目前服务器最高有32核的CPU;内存常见的单位是GB,常见的有8GB、16GB的内存,服务器最高的有64GB每条的内存;硬盘是我们最常用的存储单元,常见的硬盘大小有2TB、4TB, 10TB 这也目前硬盘的单块物理存储极限。
服务器最强大的优势是有丰富的接口和扩展性。服务器想要提高这三大硬件的配置,就是靠多,安装2-4块CPU,几十到上百条内存,硬盘组阵列,通常12块一组。什么是集群?集群就是把多台这样的服务器,装在机柜中将它们并联,将能够拆分的任务并发给每台机器让大家集体作战,这样才能够提高计算效率、减少计算时间。举个简单的例子,我们要进行100个人的宏基因组分析,如果用一台服务器就需要100天,但是我如果把这个任务分发到100台服务器同时处理,这样的话一天就可以完成了。
计算机是模拟人脑进行简单重复劳动的过程。数据存储于硬盘,读入内存用于CPU高速计算,结果再返回硬盘保存。服务器和台式机差不多,只是配置高一点,再就是数量多,将数量几台-几十台服务器并联,共同计算,这就是集群。
扩增子却是大数据时代中数据量最小的测序类型,一般配置高一点的笔记本和服务器可以搞定。价格从几千-几百万均可。研究所几百万,课题组10以内几十万,人少几万,个人初学几千就够用。

我们有了顺手的服务器,那我还有一个非常好的工作站来指挥他。推荐大家的工作站就是一台普通的笔记本外加一台扩展显示器,如果你经常做分析的话,推荐可以购买15英寸的笔记本,这样看着屏幕会大一些,同时满足我们的移动办公需求。并配一个28寸的显示器,足够大的可以将窗口分成4格,这样可以同时多任务进行管理,也方便我们平时写作、多图比较和阅读文献。

举个例子,你有一千份Excel表,主要工作是计算表格每行均值,再按结果降序排列,筛选出前3均值最高的候选。Excel中操作量与时间成正比的。编程批量操作分三个阶段:1. 手动操作几十份找规律;2. 停下工作编写程序并测试;3. 运行程序完成工作。(单位的文员最需要学编程,但他们会编程就叫数据工程师/科学家,工作效率和工资都要会成倍提高)。
如果一件事情能用手机,笔记本电脑或其他智能终端完成的时候,一定不要亲力亲为,那不叫勤奋,那叫浪费自己的生命,如果你用低效原始的方式工作,你的辛苦不值得同情。

接下来我们介绍第2部分,什么是微生物组?将带你进入真正的微生物组世界。

微生物组(Microbiota)是指研究动植物体上共生或病理的微生物生态群体。微生物组包括细菌、古菌、原生动物、真菌和病毒。研究表明其在宿主的免疫、代谢和激素等方面非常重要。近义词Microbiome即包括微生物,又包括其基因组。
上文来自维基百科,但中文是我的翻译。建议阅读原文。下图是Nature protocols杂志十年专题文章,回顾了这个领域的发展,近似的时间线展示相关领域微生物组研究开展的时间。2000年开始研究极端环境、植物、白蚁后肠、人类肠道、海洋、永久冻土、土壤沉积物等。

为什么微生物组领域这么热?近年来长期霸占着顶级杂志的封面。这篇NBT的封面是我负责的工作。这篇NG和Science是微生物所王军老师的工作。

是因为动物植物的生活环境中都充满了微生物,无论是从单细胞藻类到大型真菌、昆虫、植物、动物乃至人类的表面和内部,都充满了大量的微生物,总结一句话,脱离了微生物的生物学研究是不完整的。

我们介绍一下本领域3个最常见的词microbiota, metagenome 和microbiome。每张图代表的是相同的群体,然而不同的方法可以定义此群体可提供的不同信息。- a. 微生物群:采用16S rRNA基因测序的方法鉴定此环境中微生物的种类。- b. 宏基因组:微生物群的基因和基因组,包括质粒、强调群体的遗传学潜能。- c. 微生物组:微生物群的基因和基因组,以及微生物群的产物与宿主环境。

2001: “Microbiome”这个词才被正式提出,是随着组学发展诞生的。Microbiota是个历史悠久的词,但最近在过时,使用频率越来越低。
98年乔·汉德尔斯曼是提出宏基因组概念,耶鲁大学分子学生物学和发育生物学教授,研究土壤中的微生物和昆虫的肠道。但从90年代早期开始,她同时投身于推动女性和少数群体参与科学的运动。在奥巴马时期,他是总统的智库成员。
关于这些专业词汇的进一步学习,推荐阅读我们制作的宏基因组词汇的有声专栏,同时也推荐大家继续学习,由microbiome杂志主编编写的微生物组名词定义的评论文章。

人——HMP,2008年,1.15亿美元(刀),Rob Knight等领衔的。2016年二期5亿刀,Cutis hottenhowver等领衔。从这里我们也看出第1期主要是扩增子的研究,扩增子技术当时最成熟,而第2期主要是宏基因组数据和多组学数据,由本领域绝大多数宏基因组应用软件的开发者主导,it is a hard work。我们也同时看到,美国已经投入了6亿多刀,而中国启动的中科院微生物组计划,只投了3000万人民币,不到500万元美元,不到美国投入的1%。环境——EMP环境微生物组诗雅,Jack Gilbert领衔的。动物;植物。
10篇Nature专题报导人类微生物组计划2(iHMP)成果及展望
Nature:iHMP之“微生物组与炎症性肠病”
Nat. Med.:iHMP之“微生物组与早产”
https://en.wikipedia.org/wiki/Jo_Handelsman Jo Handelsman是Wisconsin-Madison分校的发现研究所所长,奥巴 马时代白宫科学与技术政策顾问/幕僚。最出名的著作是科学教育。

以色列技术研究所科学家Oded Beja,研究微生物与宏基因组。https://en.wikipedia.org/wiki/Oded_Beja。
Jill Banfield 加州大学伯克利,最早使用多组学;
Gordon,华盛顿大学,专注肠道研究40年,之前多次被作为诺奖的热门人选。
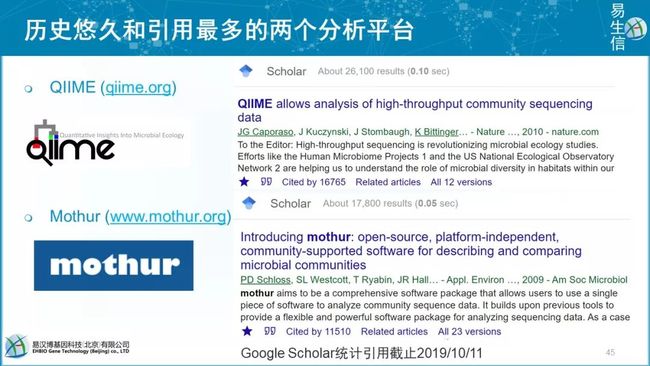
Pat Schloss发表微生物扩增子流程,引用上万。之前发布来daught和son这些零散的软件,后来打包完善为mothur,发在了3分的AEM上,引用过万。

Rob,发布了更好用分析+绘图流程QIIME,整合了200多款软件,引用近1.7万次。Jessica Green什么都研究,这是他在Ted演讲过的照片。QIIME2于16年开发,17年我参与,18年发布预印本,非正式引用近1千次。目前已经在NBT发表。

我们了解了微生物组的发展史和微生物组的基本概念看看我们。下面我们重点关注一下本次课程关注的扩增子测序的基本概念。

测序主要有以下平台。Sanger 1代,肠型的文章是测序了上千万条reads完成的,你想想成本是多少。二代测序三大平台都有产出,诺禾致源和一些医院有用Ion Torrent的,2011-2015年454使用较多,但己停产。目前90%以上用HiSeq2500, MiSeq,最新的测序平台是NovaSeq 6000;三代测序是发展趋势,但错误率太高,错误种类太任性,随机——包括插入、缺失和替换。

PacBio有10%的错误率,不仅有测错,还可能有插入、缺失等。而Illumia平均千分之一,主要是置换错误,一般长度准确。
嵌合体含量与底物DNA量,PCR循环数等有关。如30轮循环可能是5%的嵌合体,45轮时至少>15%的嵌合体;
https://bmcbioinformatics.biomedcentral.com/articles/10.1186/s12859-016-0976-y,metadata数据,很多文章缺少完整的metadata和描述文档
Microbiome杂志要求送审前就必须发布数据,也要整理好metadata表在附表中。

比对至 reference database,但不同引物差异极大,可比性不强,顶多在综述中偶尔用,研究型文章不建议使用,结果非常不可靠。读一下《引物评估》一文,你会发现不同引物对不同类型菌扩增差别极大。
整个实验最好同一条件即可。不同条件方法,DNA提取,保存方式等,可能造成假阳性结果。

培养组主要是平板、液体96孔板培养,+测序鉴定;
DNA、RNA层面二代测序;还有蛋白和代谢层面用质谱鉴定。
我们获得微生物样品,这是应该做的研究,就是培养组学,只有分离的样本,还能够有材料对细菌的功能进行研究,主要技术特长就是培养组学,这部分在国内也是比较缺少的。接下来最常用的研究层面是DNA,因为DNA的提取非常容易且质量也比较高。
DNA最常用的测序技术就是扩增子测序marker基因,常用的有16S/18S/ITS, 他们可以研究物种的多样性,但是也存在着很强烈的PCR偏好,而且也只能研究物种分类信息和丰度信息,这一技术就是我们的课的重点,他有便宜,易操作等优点,每年可以产出过万篇文章。
另一个常用的层面是宏基因组测序,该技术不仅可以获得研究对象的物种组成,还能获得对象中功能潜能的组成。但数据量和分析方法上更加复杂,每年有几千篇文章,通常为扩增子研究的进一步数据补充,可以作为独立的研究手段,这门技术涉及的分析软件与扩增子完全不同,我们在过两周还会开一门以宏基因组为主题的专题课程。
宏病毒组,是一个即要测序宏基因组,又在测序宏转录组的学科,因为病毒有DNA病毒,也有RNA病毒。无论是测序和分析都比较复杂,更要命的是病毒在研究对象中极低,有效数据比例小,需要极高的测序深度,常用靶向捕获技术来富集病毒序列。
在mRNA层面,我们研究的就是宏转录组,这与之前经常用到的转录组技术是类似的,其研究对象中的所有转录的mRNA
此外还有蛋白层面的宏蛋白组和代谢物层面的宏代谢组,他们通常也是之前研究结果的补充,而采用的设备也不同于二代测序,主要是通过质谱、高效气相或液相色谱来完成实验。

培养组学——什么菌可培养
扩增子——有什么原核生物(细菌/古菌16S)或真核生物(真菌/宿主/原生动物18S/ITS),和它们的相对丰度
宏基因组——有什么功能基因
宏转录组——哪些基因在表达
宏病毒组——有哪些种类的DNA/RNA病毒
宏蛋白组——哪些基因翻译成了蛋白
宏代谢组——有哪些种类的代谢产物
宏表观组——DNA/RNA上那些奇怪的修饰
单菌基因组——某个菌株都有什么基因
泛基因组——某种菌和亲戚间的相同与不同
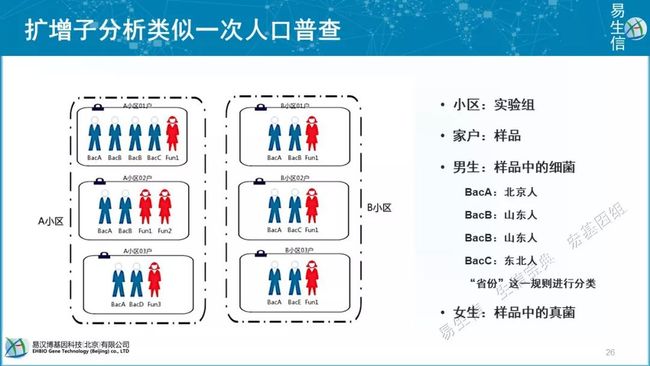
扩增子分析就像是人口普查,每个实验组相当于不同小区,每个一个样品相当于一套房间。统计人口来源,就相当于统计细菌、真菌的各类。

物种注释就相当于地址。我们为什么都是7级注释方案呢,因为我们一周有7天,7是大家最熟悉的一个数字。有谁知道,人的分类学全名7级;我们看到,人和熊猫的区别,是在目水平上的区别;人与其它灵长类,是在科水平的区别,人在科水平上已经没有朋友了,据说人类的近亲比如尼安德特人等都是被我们人类亲自消灭掉了。
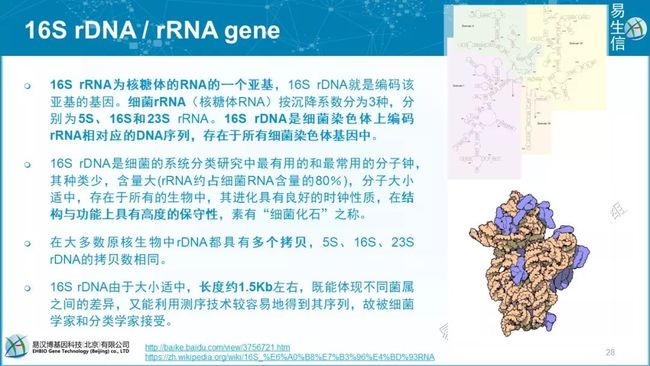
16S rDNA或16S rRNA基因,我们研究的绝对不是16S rRNA,我们扩增的是DNA。大小1.5Kb的16S长度正好可以被Sanger测通。成为首选。

所有活着的生物都有核糖体RNA。rRNAs在蛋白翻译中起至关重要的作用。rRNAs相对保守,且较少发生水平转移。
有分子钟的特征,进化分析中非常有用,用于构建生物之树。更重要的是16S rRNA 最常用,积累了大量的可用数据库,可用于分类和注释。
其实最好测全长,但技术限制 ,只能测一部分。如V4最多,还有V3-V5或V4-V5。
比如植物中也常用V5-V7
799F : AACMGGATTAGATACCCKG
1193R: ACGTCATCCCCACCTTCC
优点: 避开植物叶绿体;三V区多态性高;
缺点:存18S真菌/宿主扩增条带 ,可电泳切除

我想主要包括原生动物,真菌等,他们的测序主要采用18S作为Marker基因,真菌较多采用ITS作为标记基因,此外一些特异的类群需要特异的引物,如从枝菌根真菌,详见下面的真菌引物选择。具体的文章参见下面的文章和数据。
其实细菌也有转录间隔区,也可以有ITS引物;细菌除了我们常用的是16s之外,还可以研究一些保守的蛋白,如CPN60蛋白
病毒不能独立生活,缺少保守的基因,一般需要宏基因组+宏病毒组,也可以针对某一类研究,如T4类噬菌体Gp23、小核糖核酸病毒RdRp

最后我们总结一下扩增子分析的基本思路。
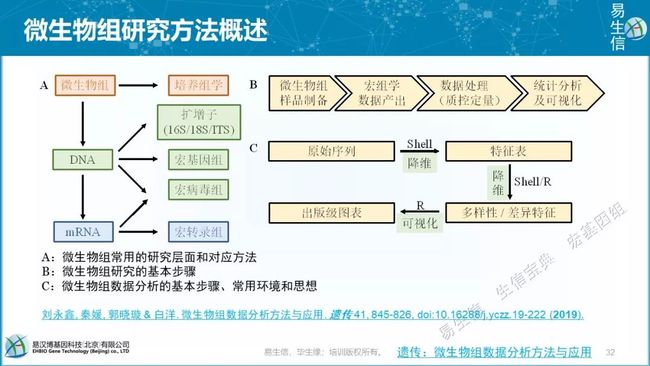
A:微生物组常用的研究层面和对应方法
B:微生物组研究的基本步骤
C:微生物组数据分析的基本步骤、常用环境和思想
可以阅读我之前发表的综述。- 遗传:微生物组数据分析方法与应用
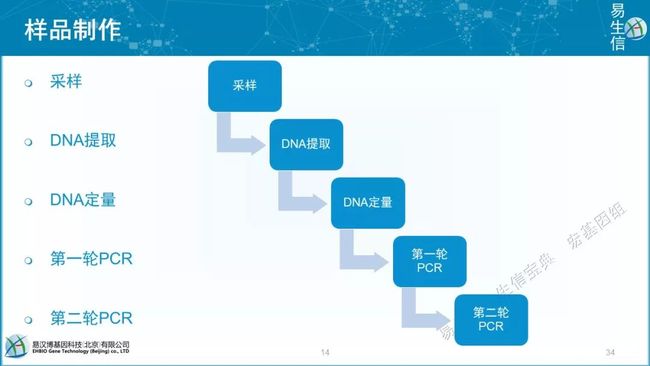
采样
DNA提取
DNA定量
第一轮PCR
第二轮PCR

2018年8月的Nature Microbiology,专门分析了污染的来源和种类。
扩增过程使用了高质量酶和超DNA free水(500/kg),减少嵌合,和试剂污染。海量测序项目,实验也要在成本和严谨间折中。
实验室:
实验室中,样品制备提取过程可引入污染
试剂可能被细菌DNA污染
在实验中加入人工混菌正对照、无底物负对照
在小样本量地尤其注意!——扩增循环越多、污染可能越严重
宿主/ 环境(宏基因组测序):
宿主在样品中残留问题
http://hmpdacc.org/doc/HumanSequenceRemoval_SOP.pdf
非目标部分(e.g. 真核细胞)可以通过细胞尺寸选择在DNA提取前去除
不需要的DNA可以使用杂交消减法移除

MiSeq 数据产量~20M paired-end(PE) 300 bp读长, 共12 Gb, 可以多达200+样本测序(按平均10万条计算),HiSeq2500单个Lane产量最高160M PE 250 bp, 80 Gb,上千样本混测,NovaSeq6000,可产出800 M PE 250 bp reads,400Gb,近万样本混测。
样品的测序量由样品的复杂度决定,简单少测,复杂多测
序列唯一的DNA barcodes加入样品中,用于区分在同一个文库中测序的样品
扩增序列具有高度同质性
需要将不同来源,或标记(marker)基因的扩增产物混合测序;或添加PhiX序列增加测序反应中的多样性
3-10万个。平均10万条,还可以测1600个样。一般最多上1000个样。才25元左右。

为什么要用两轮方法。直接在第一套中加barcodes2不行吗。我问过华大、诺禾用两步法,安诺用一步法。各有公优缺点。
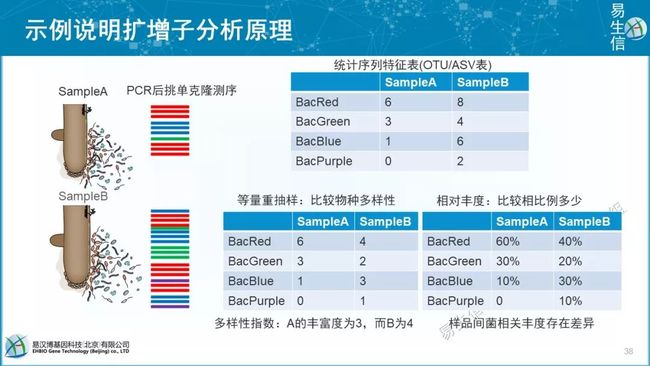
绝对丰度和相对丰度问题,spike-in, 流式,细胞计数,测OD,菌落培养法等

扩增子及常用测序获得结果为相对丰度
相对丰对的局限性
依靠内参获得绝对丰度(相对的相对丰度)

OTU(Operational Taxonomic Units)是在系统发生学研究或群体遗传学研究中,为了便于进行分析,人为给某一个分类单元(品系,种,属,分组等)设置的同一标志。在生物信息分析中,一般来说,测序得到的每一条序列来自一个菌。要了解一个样品测序结果中的菌种、菌属等数目信息,就需要对序列进行归类操作(cluster)。通过归类操作,将序列按照彼此的相似性分归为许多小组,一个小组就是一个OTU。
通常在97%的相似水平下聚类生成OTU,选择每个聚类群中最高丰度序列作为代表性序列
近期讨论发现100%更合理,即不聚类的ASV(Amplicon Sequence Variants),更容易实现跨研究比较

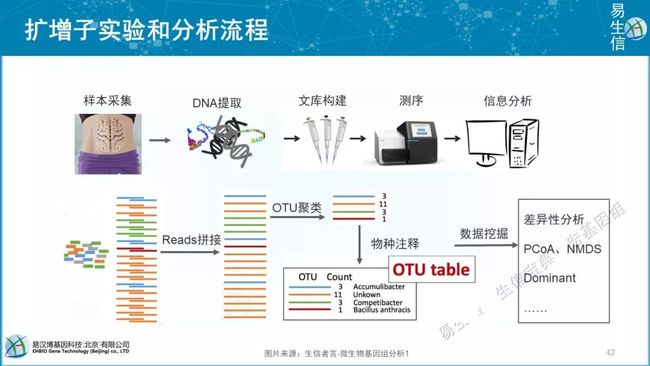


截止2019年11月27日,QIIME 17569次;Usearch 107880次;mothur 11889次,mothur由密西根大学(University of Michigan) 的Dr. Patrick Schloss领衔的团队开发的,其团队还开发有DOTUR(2005年定义OTUs和计算物种丰富度)和SONS(OTUs丰度比较)软件。详见前提提到我发表的中文综述。
数据库也各有特色,详见综述,在后面的课程中每个都会用到,物尽其用。

定量分析微生物生态;去复杂化、质控、OUT鉴定、物种分类、进化关系重建、多样性分析及可视化;它把这个领域打通了,整理了200多个软件和包,编写了150+脚本,几乎可以做本领域的任何分析。内容太多,学习成本太高,新用户无从选择。

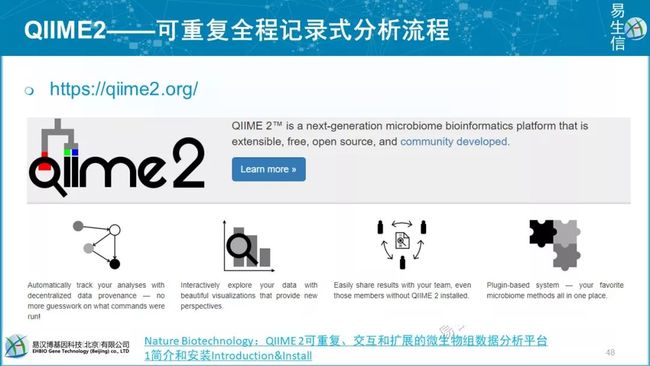
2018年由QIIME2全面接档,由Python3编写。不是升级版,而是全新的分析流程,由1的作者继续开发。格式标准化,新手体验差,适合团队强制标准化分析。

宏基因组团队参与测试和翻译QIIME2中文文档,

商用版1485$,1万多;学术版885$,6300多。
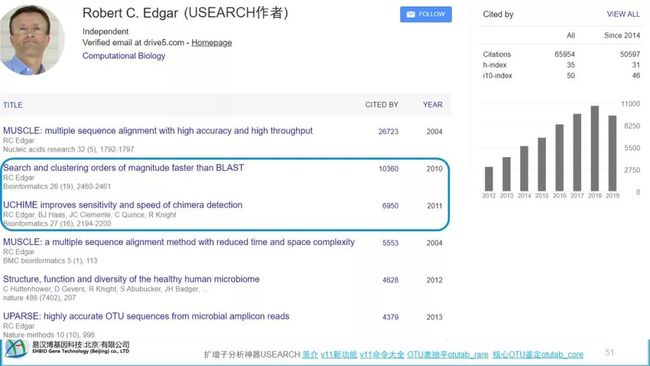
Usearch,有代表的核心算法。UCHIME和UPARSE,引用10000+,加上usearch 9725,有2万+次。QIIME和Mothur都推荐使用UPARSE聚类。
https://scholar.google.co.jp/citations?user=RzVMRc0AAAAJ&hl=en&oi=ao

一周前才只有2年前usearch8的水平,用着没有usearch10方便;3天前刚更新,功能与usearch10更接近。也有QIIME2的Plugin,学完本系统,也可以更容易上手QIIME2

https://rdp.cme.msu.edu/ RDP有整理的训练集和分类器,引用2万最多;
https://rrndb.umms.med.umich.edu/ rrnDB有16S拷贝数据收集,功能预测必须要用到,如Picrust, bugbase
https://www.arb-silva.de/ Silva是目前更新最快,最全的数据库,有很多小工具如引物评估;但缺少校正,空白和错误较多。
http://greengenes.secondgenome.com/ Greengenes是目前使用最广泛,唯一支持全面功能注释的数据库。picurst

微生物组技术:扩增子、宏基因组、宏转录组、宏蛋白组、宏代谢组
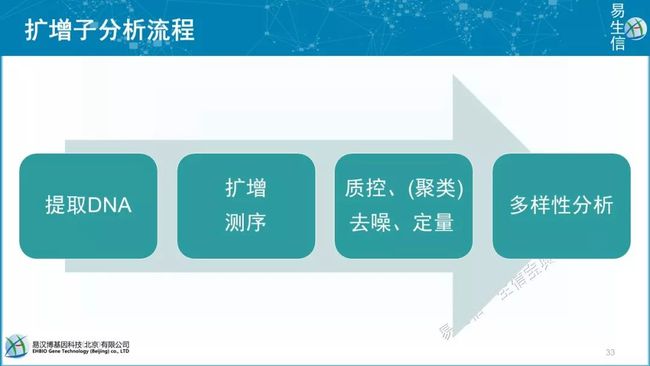
实验基本思路:取样—提DNA—第一轮PCR—第二轮PCR—测序
分析基本思路:质控—挑选代表序列OTU/ASV—物种注释—生成Feature表 —多样性分析(整体)—差异比较/机器学习(局部)
常用软件:Mothur、QIIME、USEARH、VSEARCH和QIIME2
软件选择标准:系统、配置、安装、大小、依赖关系和学习成本
数据库:GreenGene、RDP、SILVA、 rrnDB、PICRUSt和UNITE
数据库优缺点:数量、人工校正、更新时间和特色


猜你喜欢
10000+:菌群分析 宝宝与猫狗 梅毒狂想曲 提DNA发Nature Cell专刊 肠道指挥大脑
系列教程:微生物组入门 Biostar 微生物组 宏基因组
专业技能:学术图表 高分文章 生信宝典 不可或缺的人
一文读懂:宏基因组 寄生虫益处 进化树
必备技能:提问 搜索 Endnote
文献阅读 热心肠 SemanticScholar Geenmedical
扩增子分析:图表解读 分析流程 统计绘图
16S功能预测 PICRUSt FAPROTAX Bugbase Tax4Fun
在线工具:16S预测培养基 生信绘图
科研经验:云笔记 云协作 公众号
编程模板: Shell R Perl
生物科普: 肠道细菌 人体上的生命 生命大跃进 细胞暗战 人体奥秘
写在后面
为鼓励读者交流、快速解决科研困难,我们建立了“宏基因组”专业讨论群,目前己有国内外5000+ 一线科研人员加入。参与讨论,获得专业解答,欢迎分享此文至朋友圈,并扫码加主编好友带你入群,务必备注“姓名-单位-研究方向-职称/年级”。PI请明示身份,另有海内外微生物相关PI群供大佬合作交流。技术问题寻求帮助,首先阅读《如何优雅的提问》学习解决问题思路,仍未解决群内讨论,问题不私聊,帮助同行。

学习16S扩增子、宏基因组科研思路和分析实战,关注“宏基因组”
 点击阅读原文,跳转最新文章目录阅读
点击阅读原文,跳转最新文章目录阅读