ISME:二型糖尿病患者中与牙周炎相关的龈下菌群

二型糖尿病患者中牙周炎相关的龈下菌群
The ISME Journal [IF: 9.18]
DOI:https://doi.org/10.1038/s41396-019-0544-3
发表日期:2020-02-01
第一作者:Baochen Shi1
通讯作者:Huiying Li1,3
其他作者:Renate Lux2, Perry Klokkevold2, Michaela Chang2, Emma Barnard1, Susan Haake2
主要单位:
1 美国加州大学洛杉矶分校大卫格芬医学院克伦普分子成像研究所(Department of Molecular and Medical Pharmacology, Crump Institute for Molecular Imaging, David Geffen School of Medicine, University of California, Los Angeles, CA, USA)
2 美国加州大学洛杉矶分校牙学院(Section of Periodontics, School of Dentistry, University of California, Los Angeles, CA, USA)
3 美国加州大学洛杉矶分校UCLA-DOE基因组学和蛋白质组学研究所(UCLA-DOE Institute for Genomics and Proteomics, University of California, Los Angeles, CA, USA)
摘要
2型糖尿病(Type 2 diabetes mellitus, T2DM)常伴发牙周炎。龈下菌群是牙周炎发病机制中的关键因素,但其在T2DM人群中的特点还不够明确。为了更好地了解T2DM患者和非糖尿病(nondiabetic, ND)者的龈下菌群的差异,本研究对T2DM患者(n=15)和ND受试者(n=16)(两组均包括牙周健康者、牙周炎者)的龈下微生物组进行了纵向分析。采用宏基因组鸟枪法测序,研究了牙周健康、牙周炎治疗前后的菌群特点。结果发现,尽管两组的临床状况相似,ND组中牙周炎患者和牙周健康者的差异比T2DM组明显。此外,该研究发现致病菌的相对丰度不仅在牙周炎状态下高度相关,而且在T2DM的牙周健康状态下也是如此,这可能与牙周炎的进展有关。通过进一步研究龈下微生物的功能通路,确定了一组与临床状态相关的微生物标记基因。这些基因在21条通路中显著富集,其与牙周炎有关,同时也可能是T2DM和牙周炎之间的关联因子。本研究证实了T2DM患者与牙周炎相关的龈下菌群的纵向变化,提示T2DM患者更容易出现龈下菌群的生态失调,这可能是由于宿主代谢和免疫调节受损所致。
1 引言
糖尿病是一种全球发病率较高的全身性疾病。临床证据表明,2型糖尿病(T2DM)增加了患牙周炎等炎症性疾病的风险,美国有一半的成年人都患有牙周炎。糖尿病与宿主代谢失调有关,它能够上调炎症反应并促进组织破坏,而牙周炎症不利于血糖控制。炎症是糖尿病和牙周炎之间重要的关联因子。临床和动物模型研究表明,IL-1β、TNF-α、RANKL等细胞因子可能是糖尿病与牙周炎之间重要的关联因子。晚期糖基化终产物(AGEs)及其受体RAGE之间的相互作用可能会增加糖尿病患者的炎症和牙周组织破坏。最近的一项小鼠模型实验研究表明,糖尿病通过促炎细胞因子IL-17介导的炎症反应增加了口腔微生物群的致病性。糖尿病患者的免疫反应失调导致龈下微生物群失调,从而使宿主易患牙周炎。
龈下菌群在牙周炎发病机制中起关键作用。多个研究通过16S rRNA基因分析,确定了全身健康的非糖尿病(ND)受试者龈下微生物群的分类组成。这些研究揭示了健康牙周状态和牙周炎状态下的龈下菌群的显著差异,并表征了牙周治疗后菌群的纵向变化。此外,基于宏基因组鸟枪法测序分析了ND受试者龈下菌群中的潜在功能,揭示了与牙周炎相关的几个毒力因子和代谢途径。
虽然在一些病例对照研究中用16S rRNA分析了T2DM患者龈下菌群的分类组成,但缺乏治疗前后与牙周炎相关的龈下菌群的纵向分析。微生物在不同状态下的功能组成以及与ND中微生物群的比较还没有被研究过。本研究首次对T2DM患者牙周健康、牙周炎和牙周炎治疗后龈下微生物组进行了宏基因组鸟枪测序分析。确定了菌群组成和细菌间的相关性,并分析了与T2DM牙周炎相关的潜在微生物的功能通路。另外还比较了T2DM患者和ND患者不同状态下的龈下菌群特点。
2 材料与方法
2.1 患者纳入与临床检查
Subject recruitment and sample collection
对于每个参与者,在第一次就诊时都进行了初步的全口检查,以评估牙周的临床指标,包括牙龈指数GI、牙龈退缩GR、附着水平AL、牙周探诊深度PD和探诊出血BOP。每位牙周炎患者需要就诊两次。在第一次评估中,探诊深度≥5mm,牙龈指数≥1,并且在探诊时有出血,均被认为是患病位点。在第一次治疗前的第二次就诊期间,对患病位点进行采样。治疗包括牙周机械治疗(刮治和根面平整),不使用抗生素。在基础治疗后4-7周的第三次临床随访中,对至少一个牙周炎症消除的位点(PD≤4mm,探诊时无出血或其他炎症表现)重新取样进行微生物群落分析。对于每个牙周健康的受试者(PD≤4mm,BOP%≤10-15)在初次评估后的第二次就诊期间,抽取一个无探诊出血的位点进行取样分析。本研究将6个月内接受抗生素治疗或有吸烟史的受试者排除在外。大部分样本(83%)取自T2DM和ND受试者的磨牙或前磨牙部位。所有受试者在随访前都被要求在取样前48小时不要漱口,在取样前24小时不要采取任何口腔卫生措施。
T2DM和ND组牙周炎患者或牙周健康的受试者在性别上是匹配的(表1)。T2DM牙周炎患者与ND牙周炎患者之间(p=0.29)、T2DM牙周炎患者与健康牙周者之间(p=0.43)、ND牙周健康者与牙周炎患者之间(p=0.42)的平均年龄无显著性差异。但是T2MD牙周健康与ND牙周健康组年龄有显著差异(p=0.021)。对所有受试者治疗前后都进行了糖化血红蛋白(HbA1c)测定。在我们的T2DM队列中,糖化血红蛋白(HbA1c)水平在牙周治疗前后无显著性差异(p=0.36)。所有ND患者的HbA1C<6。
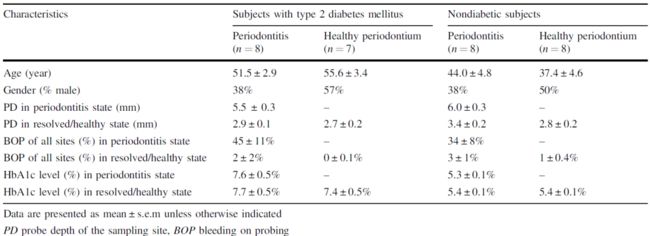
表1. 纳入人群的人口学特点和临床特点(PD,取样位点的探诊深度;BOP,探诊出血)
Table 1 Demographic and clinical data of the study participants
2.2 样本采集与DNA提取
Genomic DNA extraction
用无菌刮匙收集龈下菌斑,直接悬浮ATL缓冲液中。使用细菌裂解液处理,然后采用QIAamp微量DNA提取试剂盒提取基因组DNA。提取的基因组用EB缓冲液洗脱,短期(1周内)在−20°C保存,长期保存在−80°C。
2.3 测序与数据清理
Sequencing and data cleaning
进行鸟枪法宏基因组测序,首先使用Nextera XT DNA Library Prep试剂盒制备测序文库,然后使用100 bp成对末端读长的鸟枪法在Illumina HiSeq测序平台2000和2500对文库进行测序。在分析之前,对所有序列数据进行清理。通过Bowtie2将序列映射到人类基因组,并使用BMTagger去除人类序列。低于20的低质量碱基被修剪掉。如果两个片段中的任何一个被修剪到小于60bp或具有≥3%的不确定碱基,则该片段被移除。使用CutAdapt修剪NextEra接头和引物序列。
2.4 微生物组的分类和功能组成分析
Analyses of taxonomic and functional compositions of the microbiome
本研究采用了Schloissnig等人的方法,确定龈下微生物组的分类组成。使用Bowtie2将宏基因组序列与微生物参考基因组进行比对。为了提高的映射分辨率和效率,本研究构建了一个由124个属375个种组成的泛基因组非冗余基因组集。来自人类口腔微生物组数据库HOMD和Genbank的1309个人类口腔细菌基因组被纳入泛基因组。使用了80%的序列相似性作为阈值,并要求至少80%的基因组被测序序列所覆盖。通过计算覆盖全基因组的碱基对的数量,然后转化为物种平均基因组大小,计算出每个样本中的细菌的丰度。
使用基于基因序列相似性的BLAST法比对KEGG数据库(KO)和毒力因子数据库VFDB注释微生物功能基因。KO基因的丰度是通过将所有已鉴定物种的泛基因组中被注释为KO基因的微生物基因的碱基个数相加而以拷贝数计算的。然后根据每个样本的物b基因长度和测序深度(每108个微生p)对KO基因的相对丰度进行转化。我们用超几何分布检验确定了KO基因富集的统计学意义,并对不同临床状态的KO基因在微生物途径中的不同患病率进行了分析。如果一条途径由五个以上已鉴定的KO基因组成,则认为该途径存在。在不同临床状态下,至少有一半的样本显著富集(p<0.05)。
2.5 微生物组指标
Microbiome index
该研究使用微生物组指数来定量比较不同临床状态下的龈下微生物组。本研究将每个样本中所有已鉴定细菌的相对丰度与从ND受试者获得的数据集中细菌丰富度的平均值进行了比较。然后,根据每个细菌的统计分数,使用以下公式计算微生物群指数:

MI是微生物组指数,Xg是样本中物种(g)的相对丰度,μ1g和μ2g是来自独立数据集的处于牙周炎状态和治疗后的细菌(g)的相对丰度的平均值,tgi是在两种临床状态下比较物种相对丰度时的加权因子(g)。
2.6 统计分析
Statistical analysis
在QIIME中进行基于加权UniFrac距离和主坐标分析(PCoA)的微生物群落相似性分析。微生物群落变化的非参数多变量分析(ANOSIM)使用Mothur完成,以检验组内的微生物群相似性是否与组间的相似性显著不同。组与组之间的统计比较采用双侧分布的Wilcoxon秩和检验,纵向配对样本采用配对的Wilcoxon秩和检验。在R中使用p.adjust对多重比较p值进行校正。根据物种在样品中的相对丰度,利用Pearson相关系数计算了不同临床状态下物种间的相关关系。
3 结果
Sample and data collection
本研究招募了32名受试者,被分成四组:T2DM合并慢性牙周炎(n=8)或牙周健康(n=8),以及ND患者(n=8)慢性牙周炎或牙周健康(n=8)(表1)。对于每个牙周炎患者,在治疗前(牙周炎状态)和在治疗后(炎症消退)进行龈下菌斑样本的采集和测序。对于每个牙周健康的受试者,一个龈下样本(健康状态)被测序。所有样本均采宏基因组鸟枪法进行测序和分析。一名牙周健康的2型糖尿病受试者因为来自人体的序列较多,最终未纳入分析。最终,从47份样品中获得937亿个碱基的微生物序列,平均每个样品20亿个碱基(2 GB)。
3.1 T2DM和ND患者不同牙周状态下龈下菌群的差异
Changes in the subgingival microbiome among different periodontal states in T2DM and ND subjects
比较了T2DM和ND受试者不同牙周状态下龈下菌群的组成。本研究的数据集中共鉴定出45个菌属129个菌种。通过研究微生物与临床状态的关系发现,在T2DM和ND中,龈下微生物群在牙周炎治疗后和健康状态之间没有显著差异(图1),表明牙周炎治疗后的龈下微生物群与健康状态非常相似。ND组牙周炎状态下的龈下菌群与健康状态下的菌群有显著性差异(P<0.001),这与以前的研究结果一致,说明ND牙周炎状态下的龈下菌群与健康状态下的菌群有显著差异(P<0.05)。而T2DM患者的疾病状态与健康状态之间差异无统计学上的显著性(p=0.26)(图1)。

图1.T2DM患者与ND受试者不同牙周状态的龈下菌群比较。基于加权UniFrac距离的主坐标分析显示,T2DM和ND患者的健康状态和牙周炎治疗后之间均无显著性差异,但T2DM组中牙周健康者与牙周炎患者的微生物组差异(p=0.001)小于ND组(p<0.26)。
Fig. 1 Subgingival microbiome comparison among periodontal states in T2DM and ND subjects. Principal coordinate analysis (PCoA) based on weighted UniFrac distance shows no significant difference between the healthy state and resolved state in both T2DM and ND subjects, however, the extent of the microbiome shift from the healthy to the periodontitis state was less in T2DM (p= 0.26) compared with ND subjects (p< 0.001).
为了更好地定量比较T2DM和ND的龈下微生物群及其与牙周炎的关系,我们根据基因组序列数据制定了一个“微生物组指数”来确定龈下微生物群的状态。结果发现,ND组中牙周炎状态(中位数=0.6)与牙周炎治疗后炎症消除状态(中位数=−0.49,p=0.0069)或健康状态(中位数=−0.63,p=0.0003)之间的微生物指数有显著性差异;而T2DM组中牙周炎状态(中位数=−0.55)与牙周炎缓解状态(中位数=−0.70,p=0.382)或健康状态(中位数=−0.50,p=1)之间无显著性差异。T2DM牙周炎组与ND牙周炎组也有所不同(p=0.08)(图2)。这提示T2DM患者龈下菌群尚未完全转变为ND所见的“牙周致病性”状态,但牙周组织已出现炎症,临床表现为牙周炎症状。
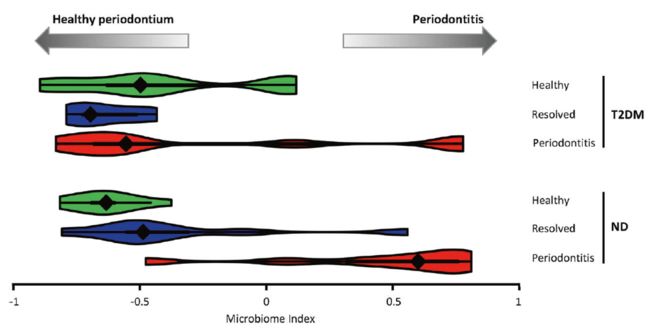
图2.微生物组指数反映了不同牙周状态以及T2DM和ND受试者之间龈下微生物组的差异。表示T2DM和ND受试者在每种临床状态下的微生物组指数。在T2DM牙周炎患者的菌群不如ND牙周炎患者龈下菌群的致病性高,而临床均表现为牙周炎的特点,这表明T2DM患者更容易受到龈下微生物紊乱的影响。
Fig.2 The microbiome index indicates subgingival microbiome differences among periodontal states and between T2DM and ND subjects. The microbiome index was represented in a violin plot in each clinical state in T2DM and ND subjects. In T2DM, the microbiome in the periodontitis state had not shifted to the extent as observed in the disease state of ND subjects, while the periodontal tissue clinically manifested signs of periodontitis, suggesting that T2DM subjects are more susceptible to shifts in the subgingival microbiome toward the disease state.
3.2 2型糖尿病患者与健康者细菌组成及相关性的差异
Differences in the taxonomic composition and correlations among bacterial species between T2DM and ND subjects
在个体龈下微生物的水平上评估了T2DM和ND受试者之间的微生物群差异。总共分析了51个平均相对丰度大于1%的细菌,它们在不同临床状态组至少两个样本中相对丰度大于1%(图3)。本研究对ND受试者的结果与之前基于16S rRNA 基因序列的研究一致,并且精确到了种水平。在ND组的牙周炎患者菌群中有15个种显著较其他两组更丰富,包括由Scransky等人定义的红色复合体的牙周致病菌(牙龈卟啉单胞菌、福赛坦氏菌、齿垢密螺旋体)和橙色复合体中潜在的条件致病菌(具核梭杆菌、直肠弯曲杆菌、中间普氏菌和变黑普氏菌),其中还包括Filifactor alocis。另外,ND组牙周健康状态下有5种物种的数量显著增加,包括黄色复合体中的链球菌(S.sanguinis、S.gordonii和S.oralis)、紫色复合体(小韦荣氏菌)和龋齿罗氏菌(Rothia Dentocariosa)。相反,在T2DM患者中,虽然检出率高的菌种与ND组表现出相似的趋势,但在相对丰度上没有统计学差异,这与上述微生物群落水平上的发现一致。

图3.不同牙周临床状态T2DM组与ND组患者龈下细菌相对丰度的比较。图中显示了T2DM和ND不同临床状态下51种细菌的平均相对丰度。在ND组中,右侧红色条带显示的15种细菌在牙周炎状态下显著多于健康状态,而绿色条带显示的5种细菌在牙周炎状态下显著低于健康状态。在T2DM组中,牙周炎和健康状态之间的差异与ND组的趋势相似,但差异没有统计学意义。
Fig. 3 Comparison of the relative abundances of the prevalent subgingival bacterial species among clinical states and between T2DM and ND subjects. The average relative
abundances of the 51 prevalent bacterial species are shown for each clinical state in T2DM and ND. In ND, 15 species indicated by the red bar on the right were significantly more abundant in periodontitis state than in the healthy state, while five species indicated by the green bar were significantly less abundant. In T2DM, the differences between
the periodontitis and healthy states had a similar trend as seen in ND, but none of the species differed with statistical significance.
牙周炎是一种多种细菌引起的疾病。基于不同牙周及糖尿病状态下细菌相对丰度的相关性,研究了细菌间潜在的相互作用(图4)。细菌复合体(complex species)在相对丰度上高度相关,在T2DM和ND组牙周炎状态下,它们都形成了一个紧密相关的细菌簇(图4a-d)。此外,在T2DM牙周健康者中(图4C),发现三种红色细菌复合体中的两种的相对丰度具有相关性,这表明与全身健康者相比,具有健康牙周的T2DM患者进展为牙周炎的风险更高。
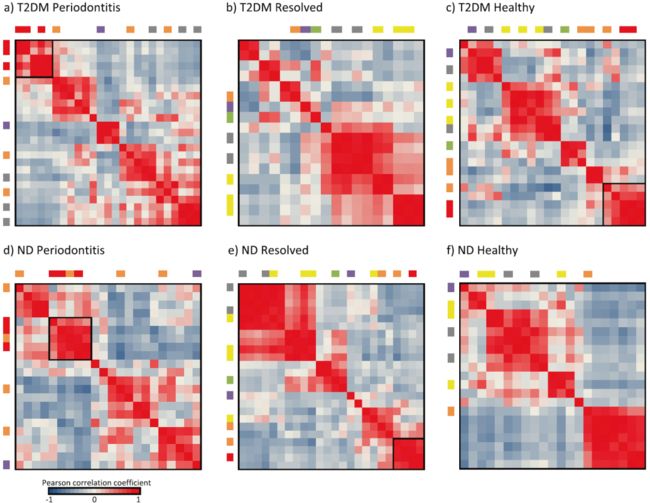
图4.龈下细菌间相对丰度的相关性
根据Pearson相关系数计算了高检出率细菌之间相对丰度的相关性。在热图中,物种在水平轴和垂直轴上以相同的顺序列出,彩色条表示基于Socransky命名的细菌复合体。热图上的黑框表示组成红色复合体的细菌簇,物种之间的平均Pearson相关系数>0.7。对相关矩阵进行层次聚类。
Fig. 4 Correlations in relative abundance between subgingival species. The correlations in relative abundance among prevalent species were calculated based on Pearson correlation coefficients. In each heatmap, the species are listed in the same order in both horizontal and vertical axes, and the colored bars indicate the bacterial complexes based on Socransky’s designation. Black boxes in the heatmaps indicate the bacterial clusters that comprise red complex species with an average Pearson correlation coefficients >0.7 between the species. Hierarchical clustering was applied to the correlation matrices.
3.3 与牙周炎和糖尿病相关的龈下菌群的功能通路
Functional potentials of the subgingival microbiome associated with periodontitis and diabetes
本研究比较了T2DM和ND患者龈下微生物组中编码的潜在功能通路及其与牙周炎的关系。共鉴定了3,201个KO基因簇,这些基因都至少有一个拷贝。549个KO基因在不同临床状态的患病率不同,标准偏差>0.2。其中,373个KO基因(67.9%)来自Socransky等人定义的代表性细菌的基因组(图5)。229个KO基因对应于特定微生物复合体,而144个KO基因来自两个或多个的微生物复合体。这些基于KO基因图谱的发现与牙周炎微生物组指数中观察到的结果是一致的(图2)。与ND相比,红色复合体的KO基因在T2DM牙周炎状态中的分布较少。另一方面,在牙周健康状态下,T2DM患者有更多橙色复合体的KO基因,这些基因通常被认为是条件致病菌。综上所述,本研究鉴定出一组指示不同牙周临床状态之间以及T2DM和ND之间微生物群差异的标记基因。
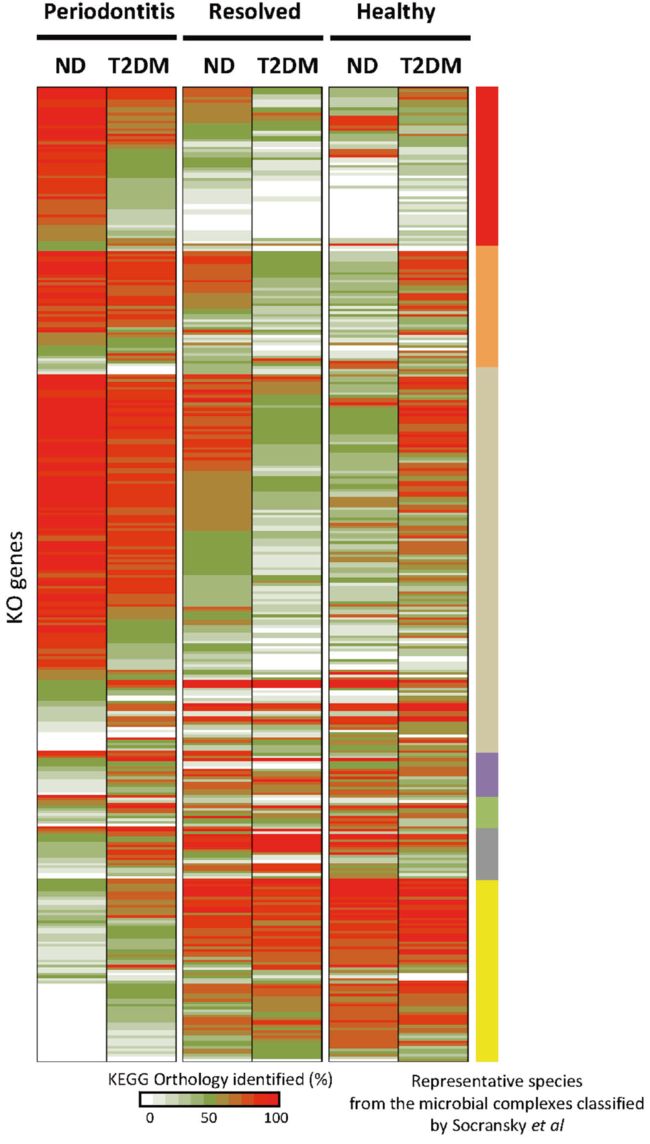
图5.KO基因在龈下菌群中的分布情况在不同牙周临床状态以及T2DM组与ND组之间的差异 共发现373个KO基因,它们在T2DM和ND组不同牙周临床状态中的检出率不同。根据Socransky等人对细菌复合体的命名,右边的色条表示KO基因所属的细菌种类。棕色标记的KO基因在该物种中是从一个以上的微生物复合体中鉴定出来的。
Fig.5 KO genes encoded in the subgingival microbiome with different prevalence among clinical states and between T2DM and ND subjects. A total of 373 KO genes with different
prevalence among the clinical states in T2DM and ND are shown. The bacterial species, which the KO genes belong to, are indicated by the color bars on the right according to
Socransky’s designation of the bacterial complexes. The KO genes labeled in brown are identified in the species from more than one microbial complex.
进一步研究了某些微生物功能途径是否与牙周炎和2型糖尿病相关,发现牙周临床状态不同患病率的KO基因在21条功能通路中显着富集(超几何分布检验)(图6)。其中,确定了与牙周炎相关的微生物途径,包括4条富含毒力因子的途径,它们在T2DM和ND的牙周炎菌群中分布更为普遍。它们是与细胞运动(细菌运动、鞭毛组装和细菌趋化)相关的通路和信号转导通路。此外,本研究还发现T2DM和ND之间微生物途径的检出率存在差异。脂肪代谢的两条途径(醚脂代谢和花生四烯酸代谢)和碳水化合物代谢的一条途径(肌醇磷酸代谢)在牙周炎状态仅在ND组出现,而在T2DM组未见。相反,T2DM牙周炎状态和健康状态下的碳水化合物代谢的3条途径(丁酸代谢、戊糖和葡萄糖醛酸相互转化、抗坏血酸和醛酸代谢)在牙周炎状态和健康状态下均明显高于ND组。
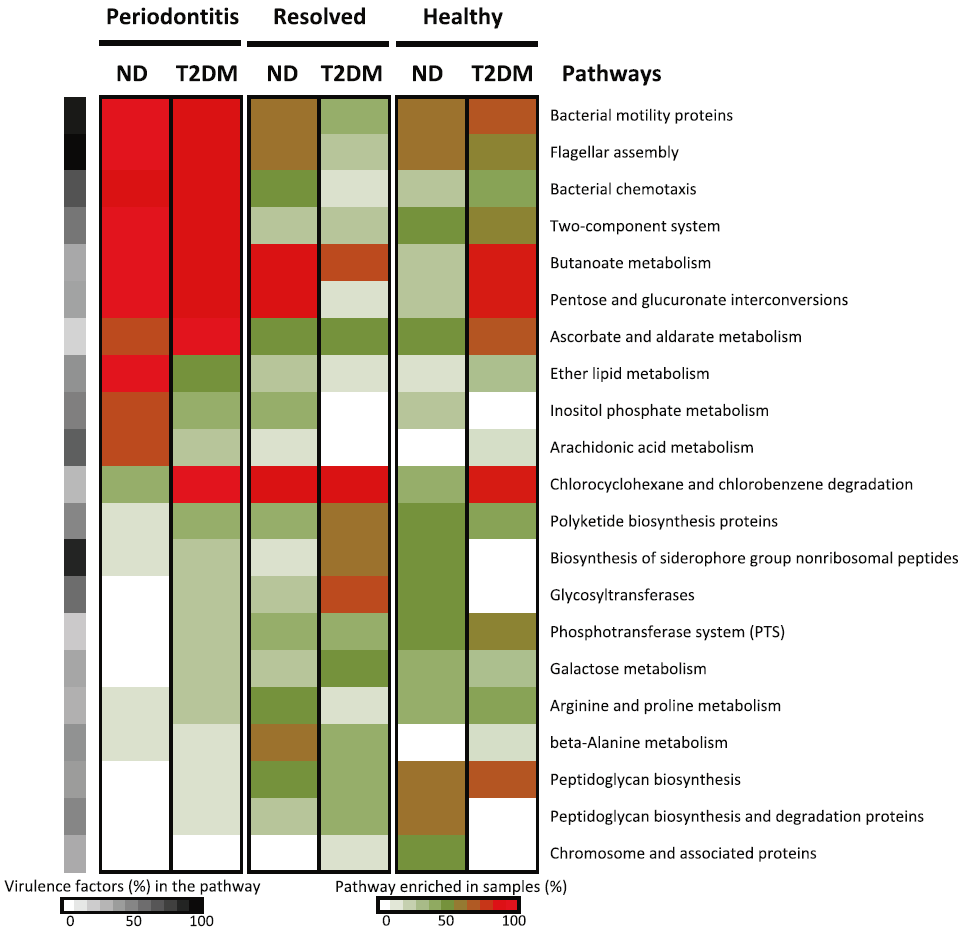
图6.与牙周炎和2型糖尿病相关的微生物途径 在21条微生物途径中,KO基因显著富集,在T2DM和ND组不同牙周临床状态中,KO基因的患病率不同。热图左侧显示了在每个途径中识别的已知毒力因子的比例。
Fig. 6 Microbial pathways associated with periodontitis and T2DM. Twenty-one microbial pathways were significantly enriched with KO genes that were different in prevalence
between clinical states in T2DM and ND. The proportion of known virulence factors identified in each pathway is shown to the left of the heatmap.
4 讨论
糖尿病和牙周炎都是世界上成年人的普遍罹患的慢性病。这两种疾病的发病机制相互关联。糖尿病增加了牙周炎的患病风险,反过来牙周炎症也对血糖控制产生不利影响。虽然高血糖与牙周炎的严重程度之间存在明显的关系,而且全身炎症状态、免疫功能、中性粒细胞活性和细胞因子生物学都与发病机制有关,但这两种疾病之间联系的机制还不是很清楚。
本研究的目的是从微生物组的角度阐明T2DM和牙周炎之间的联系。研究了T2DM患者和全身健康受试者在牙周健康、牙周炎和炎症消退状态的宏基因组水平上的龈下微生物群差异。发现两组在牙周炎状态下的龈下微生物群与健康状态下不同。利用微生物组指数,发现与ND组相比,T2DM组的微生物组从健康状态向牙周炎状态转变的程度较小。即便牙周炎的临床表现同样明显,T2DM患者的龈下菌群并没有像ND那样具有明显的紊乱特征(图2)。这表明T2DM患者对牙周致病菌的耐受性较差,牙周炎表现得更早,可能是由于宿主代谢失调和免疫反应上调引起的。本研究结果支持,在T2DM患者中更频繁地监测龈下微生物群和/或强调更严格的生物膜控制是非常有必要的,因为微生物群的微小变化可能会在糖尿病患者中引发牙周炎。
除了观察到T2DM和ND受试者之间的总体微生物组差异外,本研究还分析了与牙周炎相关的单个微生物细菌的差异。众所周知,细菌在牙齿表面的定植和生物膜的发育与牙周病密切相关。链球菌(黄色复合体)和放线菌是已知的早期定植物种,它们与牙齿表面膜中的唾液受体结合。具核梭杆菌(橙色复合体)是与包括致病性红色复合体在内的晚期定植菌相互作用的关键桥梁菌,它能够促进细菌定植和生物膜形成和演替,从而进展为牙周炎。致病性的红色复合体在牙周炎状态中都存在并且相互之间高度相关(图4),这与致病菌在牙周炎中协同作用的特点一致。比较T2DM和ND患者,发现T2DM患者在健康状态下龈下微生物菌群中橙色复合体和红色复合体的相对丰度均高于ND组(图7)。这表明T2DM患者更容易处于龈下菌群的致病状态,患牙周炎的风险更高,这与临床观察是一致的。另一方面,在牙周炎治疗后,本研究发现T2DM患者龈下微生物群落中橙色复合体和红色复合体的相对丰度均低于ND组,提示T2DM患者对牙周致病菌的耐受性较差,致病菌水平需要更低才能有效控制牙周炎症。
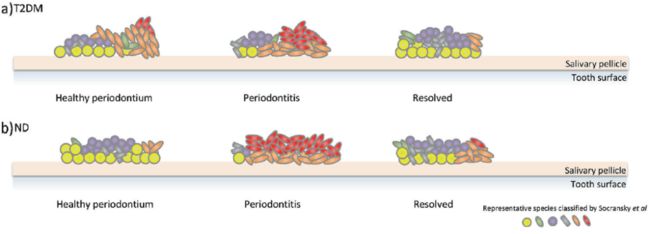
图7.T2DM和ND患者与牙周炎相关的龈下菌群特点。示意图定量地说明了T2DM和ND患者在健康状态、牙周炎和炎症消退状态下龈下微生物群组成的动态变化。与健康和疾病相关的细菌是根据Socransky等人提出的龈下复合体,以及文献中关于细菌相互作用来确定的。细菌的比例是根据本研究中它们的相对丰度来确定的。
Fig.7 Dynamics of the subgingival microbiome associated with periodontitis in T2DM and ND subjects. A schematic quantitatively illustrates the dynamic changes in the subgingival microbiome composition among the healthy state, periodontitis state, and resolved state in T2DM and ND. The health- and disease-associated species are depicted according to the subgingival complexes designated by Socransky et al. and the knowledge of bacterial interactions from the literature. The proportions of the species are scaled based on their relative abundances in our data from this study.
接下来,本研究揭示了可能将龈下微生物群与这两种疾病联系起来的微生物功能。确定了在不同牙周状态下显著丰富的21条途径(图6)。它们包括四条富含毒力因子的途径,它们在T2DM和ND的牙周炎状态中存在更为普遍。这些都是与牙周炎相关的致病途径。例如,三条已确定的细胞运动相关通路参与趋化性的细菌运动,促进牙周病原体的生长,并促进它们穿过上皮细胞层进入深层组织的定植。本研究还发现在牙周炎状态下存在一个双组分系统,它通过调节IX型分泌系统成分的基因表达,促进微生物适应宿主环境的变化,并调控毒力因子的成熟和运输。毒性因子的一个典型例子是蛋白酶牙龈素,它在牙周组织中引起炎症和组织破坏。此外,本研究还发现了三种致病途径,它们在ND牙周炎状态中更为常见,而在T2DM中则不常见。它们包括脂质代谢和肌醇磷酸代谢的两条途径,这两条途径通过脂蛋白相关磷脂酶(一组与口腔感染相关的炎性酶)联系在一起。与ND患者相比,T2DM患者在上述途径致病因子不富集的情况下就可以诱发牙周炎。
本研究还发现了有三条功能通路在T2DM组的牙周炎状态和健康状态下都富集,而在ND组无富集。其中包括参与碳水化合物代谢的途径-丁酸代谢,戊糖和葡萄糖醛酸的相互转化,以及抗坏血酸和醛酸的代谢,它们通过脱氢酶联系在一起。抗坏血酸和醛酸代谢途径与炎症疾病有关,包括牙周炎和T2DM。它的下游途径——戊糖和葡萄糖醛酸的相互转化,可能负责调节局部环境中的抗坏血酸水平。此外,本研究还发现丁酸代谢途径在T2DM患者的牙周健康状态中表达丰富,而在ND患者中没有。微生物丁酸代谢已被认为是牙周炎症的代谢标志,丁酸可以影响胰岛素敏感性。这些结果与流行病学研究一致,牙周感染对血糖控制有不利影响。因此,这些途径可能潜是牙周炎和2型糖尿病之间潜在的微生物功能关联。
样本量较小是本研究的主要局限性所在。虽然这项研究中的受试者和样本的数量与其他已发表的有关龈下微生物群的研究样本量相差不多,但未来的确还需要对更大规模的队列研究。本研究的另一个不足是T2DM和ND牙周健康组之间存在年龄差异,牙周健康的T2DM患者的平均年龄比牙周健康的ND受试者的平均年龄大(表1)。尽管如此,两组之间的微生物组成也无显著差异(p=0.189)(图2)。以往的研究未表明这个年龄范围内年龄是否影响龈下微生物群的组成。另外,虽然T2DM和ND牙周炎患者在年龄上没有显著差异(p=0.29),但本研究发现这两个疾病组之间的微生物群存在显著的差异。
5 结论
该研究首次对与牙周炎相关的高易感人群T2DM和健康受试者的龈下微生物群进行了纵向宏基因组分析。揭示了T2DM和ND在不同牙周临床状态之间的菌群差异。本研究还发现两个种群的细菌间的相关性是不同的。这两个结果都表明在进展中的牙周炎中,T2DM患者更容易受到龈下微生物失调的影响。这项研究弥补了糖尿病人群中龈下微生物群的组成和治疗后动态变化的知识空缺,糖尿病人群患牙周炎的风险更高。此外,本研究还鉴定了一组微生物基因,这些基因在与牙周炎相关的通路中富集,可能在T2DM与牙周炎之间相互关系中发挥作用。本研究从口腔微生物组的角度对T2DM和牙周炎的关系有了新的认识,未来需要更大规模的队列研究来证实。
参考文献
Baochen Shi, Renate Lux, Perry Klokkevold, Michaela Chang, Emma Barnard, Susan Haake, Huiying Li. “The Subgingival Microbiome Associated with Periodontitis in Type 2 Diabetes Mellitus.” The ISME Journal, vol. 14, no. 2, 2020, pp. 519–530. https://doi.org/10.1038/s41396-019-0544-3
责编:卢洪叶 北京大学
审核:刘永鑫 中科院
猜你喜欢
10000+:菌群分析 宝宝与猫狗 梅毒狂想曲 提DNA发Nature Cell专刊 肠道指挥大脑
系列教程:微生物组入门 Biostar 微生物组 宏基因组
专业技能:学术图表 高分文章 生信宝典 不可或缺的人
一文读懂:宏基因组 寄生虫益处 进化树
必备技能:提问 搜索 Endnote
文献阅读 热心肠 SemanticScholar Geenmedical
扩增子分析:图表解读 分析流程 统计绘图
16S功能预测 PICRUSt FAPROTAX Bugbase Tax4Fun
在线工具:16S预测培养基 生信绘图
科研经验:云笔记 云协作 公众号
编程模板: Shell R Perl
生物科普: 肠道细菌 人体上的生命 生命大跃进 细胞暗战 人体奥秘
写在后面
为鼓励读者交流、快速解决科研困难,我们建立了“宏基因组”专业讨论群,目前己有国内外5000+ 一线科研人员加入。参与讨论,获得专业解答,欢迎分享此文至朋友圈,并扫码加主编好友带你入群,务必备注“姓名-单位-研究方向-职称/年级”。PI请明示身份,另有海内外微生物相关PI群供大佬合作交流。技术问题寻求帮助,首先阅读《如何优雅的提问》学习解决问题思路,仍未解决群内讨论,问题不私聊,帮助同行。

学习16S扩增子、宏基因组科研思路和分析实战,关注“宏基因组”
 点击阅读原文,跳转最新文章目录阅读
点击阅读原文,跳转最新文章目录阅读