扩增子分析还聚OTU就真OUT了,试试unoise3
宏基因组领域是当今热门领域,也正是方法快速发展和变革的时代。之前还把 97%聚类OTU作为扩增子行业的金标准。转眼间各位大佬纷纷向OTU聚类方法拍砖,都不建议再使用。
Feature代替OTU是趋势
之前我翻译整理的QIIME2官方帮助文档——宏基因组扩增子最新分析流程QIIME2-了解分析趋势,读过的朋友会发现,里面的每个分析流程中都不再使用聚类方法生成OTU,而是调用DADA2 [1]对原始数据进行去噪,相当于以100%的相似度聚类,而仅仅对低质量序列进行去除和校正,算法识别去嵌合等;去噪的序列直接去冗余,即Feature(特征),也不再叫OTU。换了新名字,还是很不适应,本文我们还是叫OTU吧。
其实我理解,仍叫OTU问题也不大,本质上还是可操作分类单元。因为限技术和认识的限制,没有更好的方法,当前就是最好的方法。OTU和菌永远不是绝对1:1对应的,1个OTU可能包括多种菌,而一种菌也可以包括多个OTU(rDNA可以多拷贝,且拷贝间不一定完全相同)。
我理解此方法的优点主要有:
- 最大的进步是提高了种、株识别率
- 降低结果中OTU假阳性的比例;
- 有利于后续的实验和功能分析
DADA2简介
DADA2是2016年6月发表在Nature Method上[1],截止2017年10月9日Google统计引用76次。其实原理并不复杂,感兴趣的可以阅读原文,NCBI就有免费的全文。
这里只对文章中仅有的两幅图的结果简介。
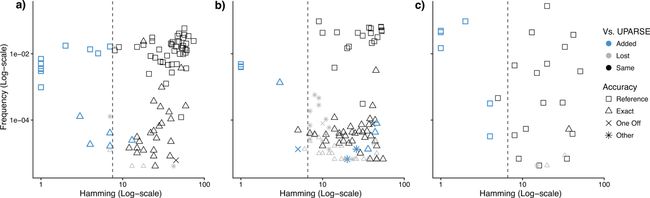
图1. DADA2[1]与UPARSE[2]结果比较。结论就是比之前的UPARSE方法更好,可以看到更多真实的OTU,进一步去除假阳性、减少假阴性结果等;
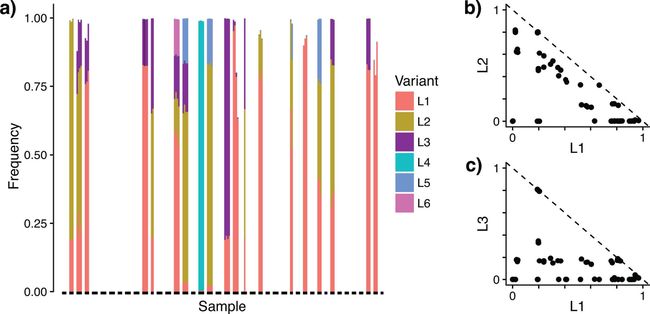
图2. 对人类怀孕期阴道卷曲乳杆菌株水平分析。可以看到6种不同的菌株不同时期的丰度变化。而之前的OTU 97%聚类是看不到株水平差异的,也是聚类最大的问题。
dada2是个R包,源码地址:https://github.com/benjjneb/dada2
安装也非常简单
source("https://bioconductor.org/biocLite.R")
biocLite("dada2")在QIIME2中是推荐的方法,可以直接调用。单独使用它,推荐阅读R包主页的帮助文档[3]。
unoise算法
看见dada2的文章孬自己的UPARSE方法不够好,Usearch大神Robert C Edgar怎能示弱,马上回家编写了unosie2 [4],10月份(dada2文章出版三个多月后)直接把unoise2算法丢在了biorxiv(预印本杂志、无需同行评审)上 https://doi.org/10.1101/081257 。原文摘要只有3句话,最后一句直接説,我的unoise2比dada2更准确(it has comparable or better accuracy than DADA2)。
文章只仅有两张图,我们简要介绍一下:

图1. unoise2的去噪原理。左图展示,高丰度的序列周围存在很多相近的低丰度序列,大部分是由于PCR和测序过程引入的。图右为去噪去后unique序列结果。

图2. 比较UNOISE2和DADA2在土壤样品中嵌合体预测的结果。结论就是説自己的算法比dada2考虑的更周到,结果更好。
原理的简介也可查看unoise的帮助页
http://www.drive5.com/usearch/manual/unoise_algo.html [5]。
unoise3生成OTU实战
上文提到dada2説比uparse更好,然后Edgar写了unoise2説比dada2更好。现在已经出了unoise3,我们当然要马上用了。
为了区别与之前97%聚类OTU的不同,作者将unoise3的结果命名为zotu。
unoise3只是usearch软件中上百个功能之一,还没用过usearch,快读读《扩增子分析神器USEARCH简介
》
文中使用所有文件下载链接:http://pan.baidu.com/s/1hs1PXcw 密码:y33d
本文的分析,是建立在扩增子分析流程基础上,想要了解每个文件的由来,请阅读《2扩增子分析流程:零基础自学-把握分析细》。
本方法是对上面链接中分析流程中第3节——聚类,和第4节——生成OTU表的另一种选择,并对结果进行简单比较。
# 97% cluster_otus聚类方法
# 聚类
./usearch10 -cluster_otus temp/seqs_unique.fa -otus temp/otus.fa -uparseout temp/uparse.txt -relabel Otu
# 生成OTU表
./usearch10 -usearch_global temp/seqs_usearch.fa -db temp/otus.fa -otutabout temp/otu_table_clusterotu.txt -strand plus -id 0.97 -threads 10
# 注:原流程中很多去嵌合、去非细菌序列的步骤是可选的,要根据具体需要是否添加。这里仅作聚类和OTU表生成的演示
# unoise3非聚类方法
# 使用unoise3非聚类直接去噪生成OTU,默认丰度阈值是reads>=8
usearch10 -unoise3 temp/seqs_unique.fa -zotus temp/zotus.fa -minsize 8
# 格式化OTU ID,不然下游分析会出现OTU表没有ID的问题
awk 'BEGIN {n=1}; />/ {print ">OTU_" n; n++} !/>/ {print}' temp/zotus.fa > result/zrep_seqs.fa
# 使用otutab mapping生成OTU表
usearch10 -otutab temp/seqs_usearch.fa \
-zotus result/zrep_seqs.fa \
-otutabout temp/otu_table_raw.txt \
-threads 9 # -id ${sim} -mapout ${temp}/zmap.txt
biom convert -i temp/otu_table_raw.txt -o temp/otu_table_raw.biom --table-type="OTU table" --to-json表1. cluster_otus与unoise3方法比较
| Method | OTU number | Mapped reads % |
|---|---|---|
| cluster_otus | 5486 | 58.7% |
| unoise3 | 4187 | 62.6% |
现在的结果是非聚类产生了更少的OTU,而可用数据比例反而增加了。
关于unoise3更详细的使用方法,可以阅读官方帮助文档[6]。
具体的好用之处,需要结合具体的科学问题来讨论吧。它们最大的优势是dada2文章图2中所示的菌株水平的分析。马上一大波基于此方法的新研究,正走在发表的道路上。
Biostack导读unoise2
在biostack上看到了一段unoise2的介绍,写的不错,同时分享给大家[7]。
UNOISE2:通过对Illumina测序平台结果错误纠正进行微生物多样性分析。Usearch主要三点: 1. 序列相似性比对, 2. 微生物多样性数据处理,逐渐构成了小生态,3. 序列处理瑞士军刀, 三点上竞争对手都很多, 第一点上 diamond 、RAPSearch等都是竞争对手,第二点 Usearch 紧随其后,另外还有 [QIIME 、Mothur 等老牌工具, 第三个问题太多了,主要有 seqtk 、 seqkit 等。 不过这个帖子提到的是新出炉的 UNOISE2,就是错误纠正(这类工具也很多), 包括了去除测序错误的序列,嵌合体序列,Phix 污染序列以及低复杂度序列等, 然后就可以直接构建 OTU表了,UNOISE2 流程推荐直接从最原始的序列开始,合并双端序列、过滤、去冗余、错误纠正、序列比对、构建OTU表、一气呵成。 另外:可以增加调整序列方向这一步,需要参考序列库,比如 RDP 的序列库,或者使用 Silva 的库。不得不提 Usearch 工具使用序列: 32位版本不管是工业界还是学术界随便用,免费, 64位版本需要进行收费了,学术界要比工业界便宜不少,现在刚进入 9.0版本,销售策略也进行了调整,从先前按年订阅, 变成现在 按大版本号订阅 ,更人性化了。
Reference
- Callahan B J, McMurdie P J, Rosen M J, et al. DADA2: high-resolution sample inference from Illumina amplicon data[J]. Nature methods, 2016, 13(7): 581-583.
- Edgar, R.C. (2013) UPARSE: Highly accurate OTU sequences from microbial amplicon reads, Nature Methods [Pubmed:23955772, dx.doi.org/10.1038/nmeth.2604].
- http://www.bioconductor.org/packages/release/bioc/html/dada2.html
- UNOISE2: Improved error-correction for Illumina 16S and ITS amplicon read. bioRxiv, 2016
- http://www.drive5.com/usearch/manual/unoise_algo.html
- http://www.drive5.com/usearch/manual/cmd_unoise3.html
- http://www.biostack.org/?p=275
猜你喜欢
- 一文读懂微生物组
- 测序分析图表解读-理解文章思路
- 扩增子分析流程-把握分析细节
- 扩增子统计绘图-冲击高分文章
- 16S预测微生物群落功能
- 岛国电影生物科普就是强—生命大跃进
- 生信媛养成记—实验汪自学生信之路-biostar handbook
- 一文读懂进化树
- 五彩进化树与热图更配-ggtree美颜进化树
- Endnote X8云同步:有网随时读文献
写在后面
为鼓励读者交流、快速解决科研困难,我们建立了“宏基因组”专业讨论群,目前己有国内几十位PI,两百多名一线科研人员加入。参与讨论,获得专业指导、问题解答,欢迎分享此文至朋友圈,并扫码加创始人好友带你入群,务必备注“姓名-单位-研究方向-职务”。技术问题寻求帮助,首先阅读如何优雅的提问学习解决问题思路,仍末解决推荐生信技能树-微生物组版块(http://www.biotrainee.com/forum-88-1.html) 发贴,并转发链接入群,问题及解答方便检索,造福后人。

学习16S扩增子、宏基因组思路和分析实战,快关注“宏基因组”,干货第一时间推送。

点击阅读原文,跳转最新文章目录阅读
https://mp.weixin.qq.com/s/5jQspEvH5_4Xmart22gjMA