Nature Method :Rob Knight发布Striped UniFrac算法轻松分析微生物组大数据
文章目录
- Striped UniFrac微生物组大尺度分析算法
- 简介
- 导读
- 正文
- 图1. 算法描述和性能评估结果
- 如何使用
- 猜你喜欢
- 写在后面

Striped UniFrac微生物组大尺度分析算法
Striped UniFrac: enabling microbiome analysis at unprecedented scale
Nature Methods, [IF 26.919], correspondence, 2018-10-30
DOI: http://dx.doi.org/10.1038/s41592-018-0187-8
第一作者:Daniel Mcdonald
通讯作者:Rob Knight
主要单位:加州大学圣地亚哥分校,儿科,计算科学与工程系,微生物组创新中心,生物工程系
大佬们都是身兼数职,一个人就挂了四个通讯单位,还好都是一个单位的。
其它作者:Yoshiki Vázquez-Baeza, David Koslicki, Jason McClelland, Nicolai Reeve, Zhenjiang Xu, Antonio Gonzalez
如果你对微生物组分析不了解,或不认识Rob Knight,一定仔细阅读下文补补基础知识
- Nature综述:大佬手把手教你分析菌群数据
简介
UniFrac是一种基于进化关系信息,计算样本间距离并进行微生物群体间的比较的方法,2005年由Rob Knight提出,截止2018年11月1日引用3762次。

此方法也在不段改进,2006推出在线工具,2010年提高计算速度,此方法最高引的4篇文章全是Rob Knight出品,共引用6609次。
近年来测序成本的下降,项目的规模越来越大,动辄成千上万的样本计算UniFrac距离进还是非常费时费力的,经常一两天没有结果,已经无法满足当前大规模数据分析的要求。
因为距离矩阵计算时,计算量和内存消耗随样本量增长呈几何积数增长;如10样品计算矩阵为100次,而1000样则为1,000,000次;时间和内存消耗增长10,000倍,对目前的计算资源是无法接受。如果能把算法优化为线性增加内存和计算量,就不再可怕了。
今天Rob Knight在Nature Method发布了改进版的UniFrac算法——条形UniFrac(Striped UniFrac,我理解Striped即为转换矩阵计算为条形向量计算之意),计算量和内存消耗仅与样本量成线性关系,完美解决了大数据计算难的问题。
导读
Rob Knight发表的基于进化距离UniFrac算法在微生物组领域使用广泛,引用超6千次;
近年来微生物组项目动辄成千上万的样本量,UniFrac算法对计算时间和内存消耗随样本量呈几何级数增长,很多大项目几乎无法完成此分析;
作者提出新的条形UniFrac方法,结果保持不变,优化计算资源消耗与样本量呈线性关系,并极大的降低内存和时间消耗,如计算EMP的2万个样本的时间可由几个月缩短至1天内;
方法提供命令行工具,并己整合至分析流程QIIME2和在线平台Qiita,方便同行使用。
正文
UniFrac度量在微生物组研究中应用广泛,但它并不适合当今的大数据集(计算过几千个样本的人,都感受过此步慢的出奇)。我们提出新算法,条形UniFrac,与原来计算结果一致,却极大的减少了内存和计算资源的消耗。同时发布了C语言共享库方便其它程序调用(https://github.com/biocore/unifrac)
UniFrac是基于进化距离的方法,进行微生物组成样本成对比较。现阶段,微生物组研究经常包括上千的样本,如地球微生物组计划(EMP)的27,751个样本,人类肠道计划的15,096个样本。现有的算法在有限的时间和空间上无法完成以上项目的分析,即使使用快速的UniFrac算法,计算EMP项目也需要几个月时间。条形UniFrac产生与现有方法一致的结果,但可提高单线程30倍的效率,同时计算量与样本数量呈线性关系,可在笔记本上24小时完成EMP项目的计算。这使科学家挖掘EMP和美国肠道计划中新的生物学观点成为可能。
为演示算法的效率,我们计算QIITA中公开的113,721个样本,采用256核计算时间小于48小时。
条形UniFrac的优势是改进了空间复杂度,通过后序遍历累计连续的度量,转换成对比较的比例向量(详细的方法、伪代码和复杂分析过程见补充笔记)。
图1. 算法描述和性能评估结果
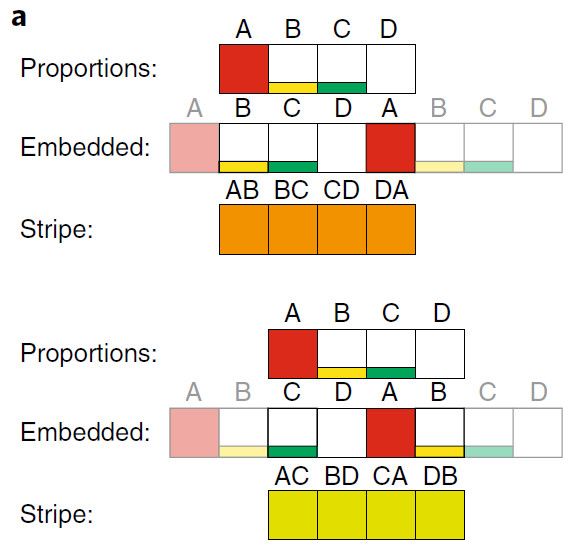
a. 进化树中某个节点,样本比例内嵌导致比例信息被复制。样本比例复制的重复模拟旋转,可以在连续的内存中计算成对的距离。
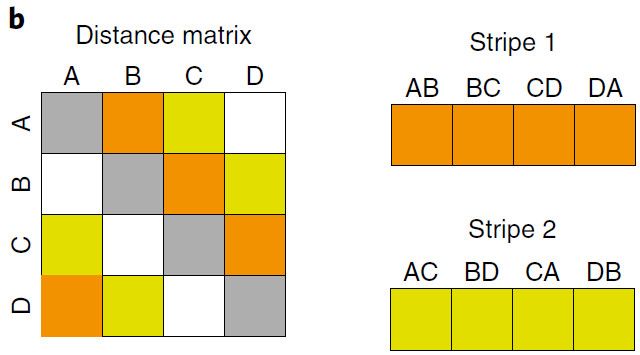
b. 四样本逻辑距离矩阵的双条形显示图;条上的标签代表成对比较,如AB代表样本A和样本B的距离。
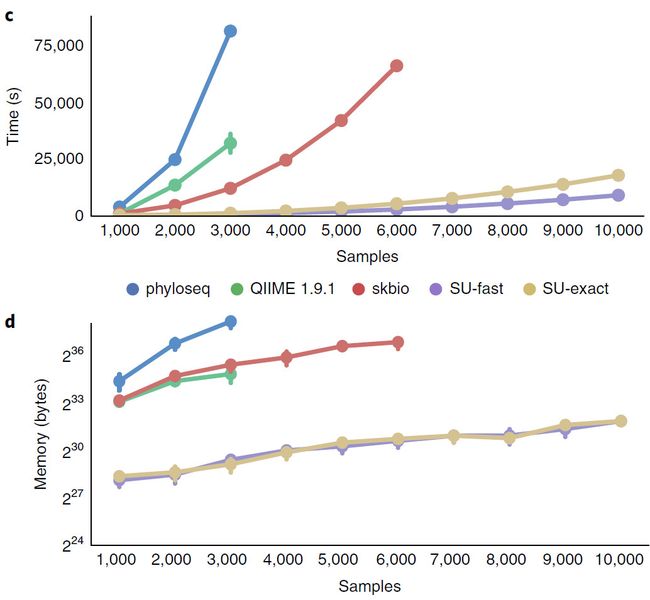
c,d, 比较有权重末标准化的UniFrac使用phyloseq, QIIME1.9.1, scikit-bio(skbio), 条形UniFrac快速模式(SU-fast), 条形Unifrac精确模式(SU-exact)经验时间©和内存(d)消耗。使用EMP 90-nt(317,314个唯一的Deblur sub-OTUs),每个数据点代表10次独立实验的均值。所有分析运行于单线程、非共享的计算结点,并且没有运行其它计算任何。任何在超过24h或大于256GB内存时被杀死。误差线表示95%置信区间。原始数据见补充数据2。
在快速UniFrac中后序聚合移除了主导的量化因子以降低空间复杂度(附图1c,d)。向量旋转(Vector rotation,在图1a表示为内嵌embedding)允许编绎器使用单说明的多数据操作(SIMD)。旋转后,成对的距离沿着对角线计算(图1b),可以使用更多缓存、任务并行和硬件层面的预读文件。同时引入了启发式搜索来忽略进化树的末端,可以减少50%的计算量(相关与准确性的计算见附图1e)。对条形UniFrac算法与现有方法比较结果见图1c,d,其性能远超当前现有方法。
如何使用
条形UniFrac算法的代码发布于GitHub (https://github.com/biocore/unifrac)。
该方法目前也已经整理到QIIME2流程中
也可以在Qiita云平台中使用。
算法也有命令行版本可用,同时提供了C语言共享库,和Python应用程序的界面。
猜你喜欢
- 10000+: 菌群分析
宝宝与猫狗 提DNA发Nature 实验分析谁对结果影响大 Cell微生物专刊 肠道指挥大脑 - 系列教程:微生物组入门 Biostar 微生物组 宏基因组
- 专业技能:生信宝典 学术图表 高分文章 不可或缺的人
- 一文读懂:宏基因组 寄生虫益处 进化树
- 必备技能:提问 搜索 Endnote
- 文献阅读 热心肠 SemanticScholar Geenmedical
- 扩增子分析:图表解读 分析流程 统计绘图
- 16S功能预测 PICRUSt FAPROTAX Bugbase Tax4Fun
- 在线工具:16S预测培养基 生信绘图
- 科研经验:云笔记 云协作 公众号
- 编程模板: Shell R Perl
- 生物科普: 肠道细菌 人体上的生命 生命大跃进 细胞暗战 人体奥秘
写在后面
为鼓励读者交流、快速解决科研困难,我们建立了“宏基因组”专业讨论群,目前己有国内外2400+ 一线科研人员加入。参与讨论,获得专业解答,欢迎分享此文至朋友圈,并扫码加主编好友带你入群,务必备注“姓名-单位-研究方向-职称/年级”。技术问题寻求帮助,首先阅读《如何优雅的提问》学习解决问题思路,仍末解决群内讨论,问题不私聊,帮助同行。

学习扩增子、宏基因组科研思路和分析实战,关注“宏基因组”


点击阅读原文,跳转最新文章目录阅读
https://mp.weixin.qq.com/s/5jQspEvH5_4Xmart22gjMA