代谢环境的大规模重建和系统发育分析(Large-scale reconstruction and phylogenetic analysis of metabolic environments)
Large-scale reconstruction and phylogenetic analysis of metabolic environments
- 摘要
- Ⅰ.引言
- Ⅱ.结果(Results)
- Ⅲ.讨论(Discussion)
- Ⅳ.材料和方法(Materials and Methods)
- 1.代谢网络和相关数据
- 2.鉴定种子化合物
- 3.协方差相关分析
- 4.系统演化分析,过渡速率和保护。
作者:Elhanan Borenstein,Martin Kupiec, Marcus W. Feldman, and Eytan Ruppin
翻译:Wendy
摘要
代谢网络的拓扑结构不仅可以为物种的代谢能力提供重要的见解,而且可以为它们进化的栖息地提供重要的信息。在这里,我们介绍了新陈代谢网络的“种子集”(基于网络拓扑结构从外部获取的化合物集)的概念,并提供了一种计算框架来计算推断给定网络的种子集。这样的种子集在代谢网络及其周围环境之间形成了生态“界面”,近似于每个物种的有效生化环境。分析了478个物种的代谢网络并确定了每个物种的种子集,我们提出了这种预测的代谢环境的全面大规模重建。种子集的组成与表征该物种环境的若干基本特性显着相关,并且与有关主要适应性的生物学观察结果一致。环境具有高度可预测性的物种(例如专性寄生虫)的种子集往往比生活在可变环境中的物种小。种子集的系统发育分析揭示了控制整个系统发育树中种子的得失的复杂动力学,以及种子和非种子化合物之间的过渡过程。我们的研究表明种子状态是短暂的,并且种子倾向于从网络中完全掉落或相对快速地变成非种子化合物。种子集还可以成功地重建系统发育的生命树。提出的“逆生态学”方法为大规模研究生物体及其栖息地之间的进化相互作用奠定了基础。
关键词: 成长环境,代谢网络,种子化合物,逆生态
Ⅰ.引言
许多生物系统都可以表示为网络的形式,可以封装它们的许多基本属性。这些生物网络的拓扑结构不仅是给出了系统中复杂的相互作用的抽象描述,而且还是系统功能和动力学的主要决定因素。特别是,已经使用了广泛的分析方法来研究代谢网络及其对各种代谢功能特性的影响,包括缩放,调节,通用性研究和对代谢基因删除的鲁棒性研究。此外,由于代谢网络在生化环境中起作用,并通过吸收或分泌各种有机和无机化合物与环境进行相互作用,因此先前的研究也解决了这些环境相互作用对代谢过程的影响。 例如,网络内部的代谢通量分布或生物体的生长速率。
但是,由于与环境的相互作用本身必须反映在进化的代谢网络的结构中,因此这些网络不仅可以用于推断代谢功能,还可以洞悉物种在其中进化的生长环境。具体来说,通过分析给定代谢网络的拓扑结构,我们表明可以识别出外源性采集的化合物(称为“种子集”;另请参见参考文献9和10)。假设一个物种的环境在很大程度上决定了它从周围环境中提取的代谢物,那么种子集就可以很好地代表其环境。因此,这种“逆生态学”方法超越了先前对代谢网络和代谢范围分析的研究,从而使追踪代谢网络和代谢生长环境的进化史成为可能。
鉴于这种方法,在本文中,我们首先介绍了代谢网络种子集的概念,并提供了一种正式的方法来计算推断给定网络的种子集。接下来,我们将该方法与大规模代谢数据相结合,以编译出一个全面的大规模数据集,以描述数百种物种的种子集。预测的种子集显示出与化合物和物种之间的生物学观察结果相符,验证了我们的计算框架和已编译数据集的潜力和相关性。然后对该数据集进行分析,以获得对代谢网络的演化动力学以及影响其与环境的界面决定因素的新颖见解。
Ⅱ.结果(Results)
我们将给定物种的代谢网络表示为有向图,该图的节点表示化合物,并且其边缘表示将底物与产品连接的反应。[具体材料和方法见附件]。这种基于图的代谢反应表示法是分析和研究代谢网络的常用工具,可以从大型跨物种数据库[如KEGG]中获得。但是,应该注意的是,这种有向图是 实际基础代谢网络的简化,而忽略了例如反应化学计量(请参见讨论)。网络中出现的化合物称为发生的化合物。形式上,我们将网络的种子集定义为 不能由网络中的其他化合物合成(因此是外源获得的)且其存在是允许网络中所有其他化合物的产生的最小子集(图1A)。
我们对种子化合物的定义与基本化合物的定义不同,因为我们要求无论它们在给定环境条件下的实际动态激活如何,网络中所有化合物的生产(以及所有代谢途径的潜在激活状态)。在实践中,生物可以在广泛的环境条件下生存,并且在每种环境中,可以使用一组不同的外源获得的化合物来激活网络中仅一部分途径。因此,种子集可以被认为是所有这些环境中所需的基本集的并集。 假设各种替代途径已经进化并由于在某些环境中的适应性价值而得以保留,种子集代表了总体的静态代谢“界面”(或代谢“潜力” )(可以用作其有效生化栖息地的特征代表)。
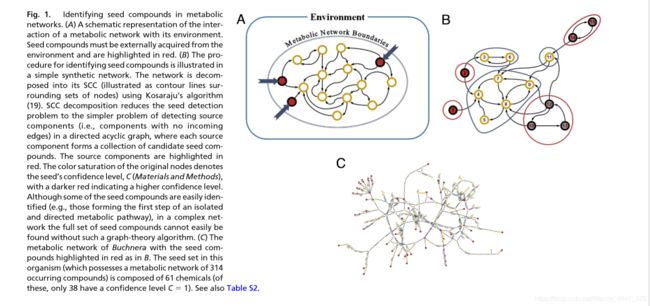
我们设计了一种基于图的算法来检测给定网络的种子集(有关更多详细信息,请参见附件及图1 B)。该算法基于一种用于 强连接成分(SCC)分解 的快速方法,因此可以轻松扩展并应用于大规模网络数据。接下来,我们使用数据构建了478个物种的代谢网络(表S1)。 从大型代谢反应数据库(见附件)中提取数据,并应用种子集检测算法来识别每个网络中的种子化合物(数据集S1)。该汇编产生了数百个物种的预测生化环境的全面大规模数据集,并有助于此类种子集的种间比较。
显然,大规模代谢数据通常基于基因组注释,主要来自基于比较的自动化方法,因此注定是不完整和不准确的。这可能会对推断的种子集的组成产生显着影响。但是,检查丢失或错误数据的影响(SI文本和图S1),我们发现所标识的种子集对原始代谢数据的扰动相当鲁棒。 不过,考虑到与这些数据相关的固有噪声和不完整性,我们在这里主要集中在识别重要的大规模统计信号和系统进化模式上,这些信号表征整个生命树中种子集的组成。
为了举例说明通过我们的分析获得的种子集的组成,并将其与有关生物体环境的已知发现进行对比,我们首先关注栖息地简单且特征明确的单个物种。专性的胞内共生蚜虫蚜虫丧失了许多生物合成基因,并证明了其与蚜虫宿主的共生极为成功。 它为寄主提供了蚜虫必不可少的氨基酸(即,蚜虫无法合成),并依靠寄主提供了无法合成的营养物质(例如非必需的某些氨基酸)。布氏菌仅对葡萄糖和甘露醇保留底物特异性转运蛋白,并负责硫酸盐同化作用,这是蚜虫所不具备的能力。最后,除编码2-氧戊二酸脱氢酶复合物的那些基因外,它缺乏所有的TCA循环基因。 通过我们的分析获得的蚜虫双歧杆菌种子组的组成(图1C和表S2)与上述观察结果很一致。它含有蚜虫,谷氨酸和谷氨酰胺中最丰富的非必需氨基酸[Buchnera将其用作合成其他必需氨基酸的底物],并且不含所有宿主必需氨基酸。种子还包括葡萄糖和甘露醇(作为唯一的碳源),2-氧戊二酸酯和硫酸盐,以及硫胺素(维生素B1)和精胺(必需的生长因子)。
为了进一步确认通过我们的分析获得的种子集的组成与有关各种物种从其环境中提取的化合物的已知大规模生物学观察结果一致,在我们的分析中,我们考虑了几种关键化合物并检查了它们在存在的化合物中的存在和不存在模式, 所有物种的种子集(系统发生模式)和种子集(系统种子模式)(SI材料和方法以及图S2)。例如,虽然许多物种可以合成它们所需的所有氨基酸,但动物已经丧失了制造某些氨基酸(称为必需氨基酸)并通过饮食获得它们的能力。相反,一些专性的细胞内寄生虫丧失了产生非必需氨基酸的能力,并依靠它们的宿主来外源提供这些氨基酸。比较苯丙氨酸(一种必需氨基酸)和谷氨酸(一种非必需氨基酸)所得到的系统发育模式,我们发现这些模式与上述观察结果完全一致(图S3)。另一个例子是生物素(维生素B7),它是羧化反应中必不可少的辅助因子。在最近的比较基因组研究中报道的42种物种中,它们可以合成生物素(因此不需要从环境中吸收生物素),实际上,有40种具有生物素的存在性化合物而不是种子化合物。在据报道缺乏这种能力的24种物种中,有20种确实具有生物素作为种子。 有趣的是,这四个似乎缺乏合成生物素的能力且在种子集中没有生物素的物种,都具有相同的生物素生物合成前体,即脱硫生物素,已被证明可以使多种生物素营养缺陷型细菌在种子中生长, 缺乏生物素。其他示例和细节在《附件》中提供。鉴定出的种子也与整体拓扑特征相关,并富含某些代谢途径(附件文本)。
涉及多种物种和多种化合物的进一步验证建立在有关人类埃希氏菌病(一种新兴的传染病,主要通过壁虱或吸虫)传播的几种药物的生物合成能力的数据上。这些细胞内媒介传播的病原体在我们的工作中特别有意义,因为它们的生命周期涉及脊椎动物和无脊椎动物宿主,因此需要代谢适应非常不同的环境(另请参见参考文献28)。最近的一项比较基因组学研究探索了这些新测序的无性科(以及立克次体和其他昆虫共生体的其他物种)合成氨基酸,主要维生素和辅因子的能力(参考文献29和表5)。将我们确定的种子集与这些数据进行比较(跨度物种和30种化合物;表S3),我们检查了参考文献中是否报告了化合物。 29种不会在特定物种中合成的(因此是外源获得的)被我们的算法正确分类为种子,并且可以合成的化合物是否正确分类为非种子(见附件)。我们发现两个数据集之间的总体一致性很强,分类精度为79%[P <10 -6; 10个辅助因子的准确度和准确度为93%(P<10 -5) [见附件]。此外,仅关注我们正确预测外源获得的种子化合物的能力,我们就可以达到95%的准确度(所有预测的种子中正确识别的种子的百分比; 见附件)和67%的召回率(正确识别的种子落入外源获得的化合物的百分比)。 考虑到基础代谢数据中固有的噪声(请参见SI Text),这些分数证明了我们方法成功识别外源性获得的化合物的能力。
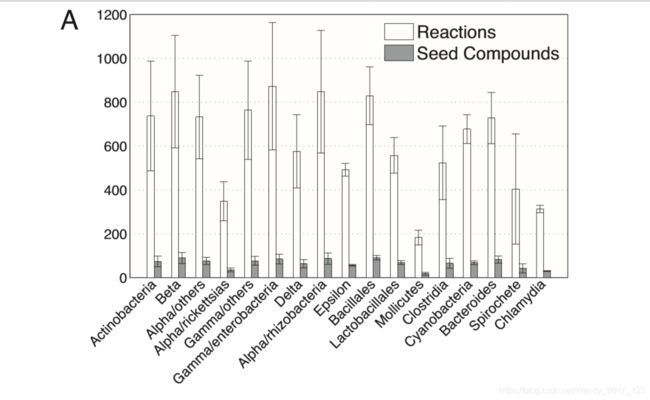
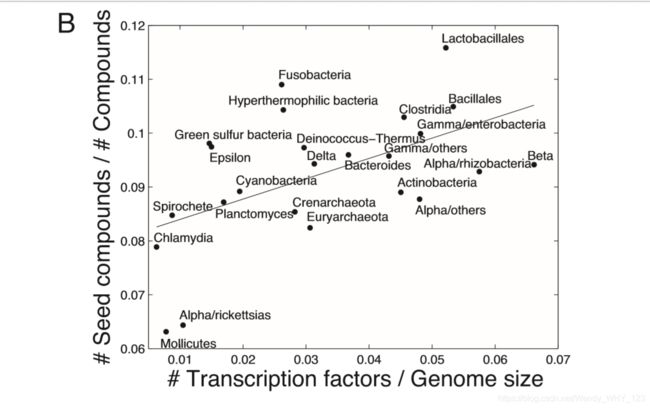
在证明获得的种子集数据与化合物和物种间的各种生物学观察结果一致之后,我们现在转向确定所鉴定种子集的大小和组成与物种环境之间的大规模关系。在这里,我们仅将分析局限于原核物种,可以获取大规模的环境数据(见附件)。此外,原核生物促进了不同生境之间的比较,而没有与组织特异性代谢物营养变化的营养水平相关的并发症。 我们发现,生活在极端狭窄的生境中的生物(例如,Archaea)倾向于拥有较小的网络和较小的种子集(另请参见参考资料13和图S4)。生物体环境与网络结构和组织之间的这种强相关性尤其表现在细菌门上;具有最小代谢网络和最小种子集的门菌是立克次体,毛囊菌,螺旋体和衣原体,它们大多是专性的细胞内寄生虫,栖息于明确且可预测的环境中(图 2A)。此外,生活在高度可预测的环境(例如,海洋热风口)中的专门物种不仅比生活在多个栖息地中的物种倾向于在其网络中出现较少的化合物,而且还需要这些化合物的分馏分数较小(P<3×10-4和P <0.03 ,分别; Wilcoxon 秩和检验)。可变环境与较大种子集之间的正相关性进一步得到了包括种子集在内的所占化合物分数(种子集大小的归一化度量,控制网络大小变化)与环境变异性指数(0.27,P<0.004; Spearman等级)之间显着的统计相关性的进一步证实。 相关性;材料和方法)。此外,我们还在种子集中的化合物含量与转录因子数量与基因组大小之间的比率之间发现高度相关性(0.4,P<2×10-7; Spearman等级相关性;材料和方法以及图2B), 后者与生境变异性相关。这些结果表明,尽管依赖高度可预测的环境的物种可以从中吸收许多化合物(而不是合成它们),但总的来说,与必须在各种环境小生境中生存的生物相比,它们提取的化合物要少得多。 这些发现也支持我们的直觉。 种子集是对应于该物种可以生存的不同环境条件的各种基本集的组合。

使用变异相关分析(见附件),我们进一步确认了各种原核生物的生长环境不仅与种子集的大小相关,而且与它们的组成相关。关于每个物种的生长环境的数据用四个属性(盐度,氧气需求,温度范围和栖息地)的矢量表示,使用离散类别描述每个属性(见附件)。考虑到可以获得环境数据的446个细菌和古细菌类群,在环境的“签名”和种子的种子组成之间发现0.25的显着相关性(Pearson相关检验,P<10-3;见附件) 一种物种。尽管存在的集明显大于种子集并且可能携带更多的信息,但该相关实际上高于环境特征和发生的化合物组成之间的相关性(0.21,Pearson相关检验,P<10-3)。对于某些环境属性,具有相同属性值的物种也趋向于具有相似的种子集。特别是,具有微需氧,兼性和有氧需氧量,具有多个生境和中温温度范围的物种具有比预期偶然得多的相似种子集(经过多次测试校正的P 0.05;见附件)。
接下来,我们描述了进化动力学,这些进化动力学控制着整个系统发育树中种子和非种子化合物的得失,以及种子与非种子状态之间的过渡。 为此,我们使用分子进化分析(31,32)和基因保守性分析(33)借用的方法并使用几种系统发育分析方法(包括最大简约算法和最大似然模型;见附件)来分析seedoc文档集。 具体而言,我们 考虑了基于参考序列的树,并估计了各种化合物整合到进化的代谢网络中(和从中丢失)的速率,以及种子化合物变成非种子的化合物的速率,反之亦然(材料和方法) ; 表格1)。这些估计控制着系统发生关系,可以将物种动态与过渡事件分开。
我们的发现表明,新化合物以种子或非种子化合物的形式集成到代谢网络中,其中非种子的整合事件的频率是种子的整合的1.5–2倍(表1)。然而,种子化合物比非种子化合物具有更高的趋势,可以完全从网络上掉下来。而且,所合成的化合物在可以看到的化合物上的转化速率要比逆过程高(表1)。这些发现表明,一般而言,种子状态是化合物“生命”中的一个过渡阶段,种子化合物往往从网络中完全掉下来,或者相对较快地转变为非种子化合物。 我们进一步计算了几种保护措施(见附件),以估计种子和非种子状态的预期进化“寿命”。 我们发现种子状态的保守性明显低于非种子状态(丢失率分别为0.0523和0.0378; P<10-12,Wilcoxon秩和检验),再次确认了 种子状态的瞬时性质。
最后,我们分析中所包括的各种物种的种子含量以类似于基于基因含量的系统发育学的方式用于重建系统发育树(见附件)。值得注意的是,这种生命树不仅成功地将大多数生物分类群聚在一起(图3),而且与基于整个生命周期的树一样准确(通过其与基于参考序列的树之间的距离进行测量; 见附件)。 尽管基于化合物的数量要少得多(表2)(种子化合物平均仅占化合物的10.8%)。这种偶然发生的化合物的小子集不会偶然地预测到这种准确的重构,如随机化合物集获得的明显不那么准确的树所证明的(表2),这表明所 鉴定的种子集是每种物种及其进化史的重要和基本特征。主成分分析进一步表明,种子集的组成可用于划分主要分类组(SI文本)。

Ⅲ.讨论(Discussion)
这项研究通过对478个代谢网络(和2200种代谢化合物)进行跨物种分析,推断出每个物种从其环境中提取的化合物,从而对代谢环境进行了大规模重建。我们的分析仅基于网络拓扑,而忽略了代谢反应的许多其他特性,例如化学计量(反应物与每个反应产物之间的定量关系),速率和动力学。网络表示也权衡所有路径,而忽略分解代谢和合成代谢途径之间的重要区别。 将这些特性整合到代谢网络模型中并应用更复杂的分析(例如基于约束的化学计量模型)可能会产生更多的准确结果(另请参见SIText,了解基于拓扑的分析对自催化循环中种子鉴定的潜在影响)。因此,使用我们简化的网络模型获得的种子集可能会在代谢数据中暗示大规模模式,而不是反映准确的化学计量约束。 然而,尽管存在这些缺点,但拓扑分析仍具有几个重要的优点:对于此处介绍的分析类型而言,最重要且必不可少的是,可以轻松获得数百种物种的代谢网络拓扑(不同于仅对少数物种可用的化学计量模型和动力学模型),这使得系统发育可以进行大规模分析。基于拓扑的模型也适用于方法和算法(大多数是从图论或复杂的网络分析中借用的),从而简化了其他复杂模型中难以处理的分析。 具体而言,种子集识别(我们已在图形表示中引入了一种快速且相对简单的算法)在化学计量网络中是一项极富挑战性的任务,需要复杂的优化方案(例如混合整数编程),而这些方案无法扩展到实际规模的网络。最后,已确定的种子化合物数据集与物种和交叉化合物之间的生物学观察结果不一致,并促进了影响种子集的各种环境因素的表征以及代谢网络演化的动态。

种子集的大小显示出与环境可变性紧密相关,并且显示出它们的组成具有多种环境特征。估计的种子和非种子之间的过渡速率以及化合物的得失率提供了控制网络进化的总体模式的详细特征,并提出了复杂的动态过程。化合物的种子状态似乎是相对短暂的,而此类化合物往往会迅速丢失或转化酮种子化合物(很可能会发生适应,并且会从新种子合成这些化合物)。这些水平基因转移的动态选择研究表明,这种转移更有可能发生在涉及营养吸收或最初代谢步骤的周围反应中。种子向非种子的转化率高于逆向过程的转化率,也与网络进化的逆向模型 一致(假设基质驱动的过程,代谢途径向后组装,代谢途径相反) 途径)。但是,非种子化合物的整体整合率较高,非种子向种子的转化率仍然相对较高(代表外部获取以前生产的化合物的策略的演变)表明其他过程[例如,拼凑模型(36)]可能起着重要作用。 在代谢网络的进化中起重要作用,并且这些过程不是相互排斥的(另见参考文献37)。成功地重建基于种子的生命树,进一步体现了其对各种环境生态位的出色适应能力。本文介绍的种子集分析和上述发现说明了“逆向生态学”方法的巨大潜力,并促进了有关进化力的进一步大规模,跨物种研究,这些进化力决定了 生物体及其生物环境之间的相互作用。
Ⅳ.材料和方法(Materials and Methods)
1.代谢网络和相关数据
代谢网络数据是从KEGG数据库(15),版本41.1(2007年2月)下载的。 总共重建了涵盖所有生物分类群的558种(表S1)的代谢网络(SI材料和方法)。分析中排除了基因组草稿和EST重叠群(KEGG生物编码,带有前缀“ d”或“ e”)。我们还丢弃了具有100个反应的物种,总共保留了478个物种。 还重建了一个由反应物种联盟组成的附加网络,并将其称为全球网络。有关细菌和古细菌环境的数据可从NCBI基因组计划的原核属性表中获得(www.ncbi.nlm.nih.gov/genomes/lproks.cgi)。每种物种都代表了其盐度要求(非嗜盐,中度嗜盐或极度嗜盐)的载体特性,其需氧量(需氧,微需氧,兼性或厌氧),其温度范围(低温,嗜冷,嗜温,嗜热,或适应多个温度)。有关详细说明,请参见《附件》。上表的基于进一步手动版本化的其他环境数据以及对117种细菌物种的环境变异性进行的排名是从参考文献30中获得的。 我们还从参考文献中获得了有关159种细菌的转录因子数量和基因组大小的数据。并使用这两个值的比率作为环境变化的定量补充指标。
2.鉴定种子化合物
使用Kosaraju的算法(19)(SI附件)将每个网络分解成其紧密连接的组件。牢固连接的组件具有最大的节点集,因此u和v的每对节点都有从u到v的路径以及从v到u的路径。紧密连接的组件形成一个直接的循环图,该图中的节点是该组件的节点,而其边缘是连接两个不同组件的节点中的原始边。在此图中,每个组件都没有传入边且至少有一个出站边被定义为源组件。SCC分解中的每个源组件都是候选种子化合物的集合。种子化合物的集合必须确切地包含每个来源成分中的一种化合物,并且不应包括任何其他化合物。 在下面,网络提供直觉(基于网络的图表示;另请参见上面有关反应化学计量的讨论):首先,应该注意的是,每个强连接的组件都是等价类; 如果可以生产组分中的一种化合物,则所有其他化合物都可以生产。其次,由于 源成分没有任何进入的边缘,如果源成分中没有化合物存在于种子集中,则此成分中的任何化合物都不会产生。最后,如果种子集合中包括来自每个源成分的脂肪族化合物,则可以找到从种子化合物到网络中任何其他化合物的路径,因此可以生成网络中的所有化合物。由于所有化合物都可能包含在种子集中,因此为每种化合物指定了置信度水平[C =1 /(组分大小)],表示该化合物被诱捕的可能性。我们使用C≥0.2的阈值来确定是否应严格禁止使用一种化合物(包括属于5号或更小尺寸来源成分的所有化合物)。使用此阈值,我们平均仅丢弃3.3%的种子。 使用其他阈值(特别是C≥0.1或C≥0.01)不会显着改变任何结果。 数据集S1描述了每个物种中种子集的组成(具有关联的C值)。另请参见图S6,该图说明了酵母的代谢网络,并突出了种子化合物。
3.协方差相关分析
为了 检查种子集组成与所有细菌物种的环境特性之间的相关性, 我们采用了一种与参考文献 39中使用的类似的测定方法。给定N个物种,将重建两个N×N个距离矩阵Ss和Sh。Ss表示各个物种的种子集之间的成对杰卡德距离。Sh表示描述这些物种环境的属性向量之间的成对汉明距离。计算了形成Ss和Sh的下三角的(n2-n)/ 2个条目之间的Pearson相关性。通过将该物种的标签标记1,000次并计算概率以达到等于或更高的绝对值相关性分数,计算了所得相关性的统计显着性。另一种测定方法检查是 具有特定环境属性值的各种物种的种子集之间的相似性。 计算具有该特定属性值的种子集秋天物种之间的平均成对距离,并将其与100,000个相同大小物种的随机集合所获得的平均距离进行比较,以确定其统计意义。通过错误发现率程序校正了所得的P值,以进行多次测试。
4.系统演化分析,过渡速率和保护。
我们认为一个建立良好的,基于序列相似性的树作为参考系统发育树。该树基于31个直系同源物,包括相对大量的物种,涵盖了可获得代谢数据的大多数分类组。我们基于系统发育的分析仅限于可与参考树匹配的物种,导致总共178个物种。每个物种(既有物种或祖先物种)中的给定化合物可以采用以下三种不同状态之一:不存在(完全不存在于出现的化合物集中),非种子(不属于种子集中的出现的化合物)或种子( 这是种子集的一部分)。我们的种子分析确定了现存物种中每种化合物的状态。为了计算整个系统发生树上不同状态之间的进化转变速率,我们应用了三种分析方法,将果寡糖使用二核苷酸取代(参见SI附件)。在第一个分析中,使用 Fitch的小简约算法 预测了化合物在系统发生树的每个内部节点(代表祖先物种)中的状态。Fitch的小简约算法为系统树的所有内部节点找到了最简约的状态分配,给定了一种将状态分配给当前物种节点的系统模式。然后,我们根据Tamura和Nei方法(一种用于替代率估算的常用方法)(SI附件)来计算不同状态之间的替代相对频率。在第二种分析中,估计了树的内部节点中每种化合物的状态,但这一次是基于Kreimeretal(28)的代谢网络潜在物种的重建。这些集合检测算法适用于每个星形网络,以获取内部节点中正在发生的化合物和种子化合物的集合。Tamura和Nei的方法再次用于估算相对转换率。在第三次分析中,将最大似然方法应用于所有化合物的系统发育模式,以获得取代率的最大似然估计。这是通过使用UNREST模型的PAML程序包计算。两种附加的保护措施,即遗传损失倾向(PGL;最大似然度量)和遗传损失率(GLR;最大似然度量),还应用于各种化合物的分子式模式,以计算化合物失去其作为种子化合物的状态的趋势(其状态为非种子复合过程)。 通过这些措施,我们不会在化合物从网络完全掉落的情况和状态已转换为另一种状态的情况之间区别不大,而只是要刻画每个状态的保护水平。因此,可以将这些保护措施视为代表该州的平均“寿命”。 从PGL量度获得的结果在质量上与通过GLR量度获得的结果相似,因此未列出。