ChIP-seq(2):ChIP-seq peaks可视化(Rstudio) 学习笔记
好了,可以在R语言里玩耍了。
参考资料:
ChIP分析流程
Chip-seq 实战分析流程
1.安装软件
(之前安装过,先检测一下)
source ("https://bioconductor.org/biocLite.R")
biocLite("ChIPseeker")
biocLite("TxDb.Mmusculus.UCSC.mm10.knownGene")
biocLite("org.Mm.eg.db")
biocLite("clusterProfiler")在安装 org.Mm.eg.db 时,出了一些问题,需要安装所对应的包,比如我这个报错, AnnotationDbi 、rlang 需要安装**,我也是捣鼓了2小时才发现原来是个样子。
BioC_mirror: https://bioconductor.org
Using Bioconductor 3.7 (BiocInstaller 1.30.0), R 3.5.1 (2018-07-02).
Installing package(s) ‘org.Mm.eg.db’
installing the source package ‘org.Mm.eg.db’
trying URL 'https://bioconductor.org/packages/3.7/data/annotation/src/contrib/org.Mm.eg.db_3.6.0.tar.gz'
Content type 'application/x-gzip' length 69432647 bytes (66.2 MB)
downloaded 66.2 MB
* installing *source* package 'org.Mm.eg.db' ...
** R
** inst
** byte-compile and prepare package for lazy loading
**### AnnotationDbi 、rlang 需要安装**
Error: package or namespace load failed for 'AnnotationDbi' in loadNamespace(i, c(lib.loc, .libPaths()), versionCheck = vI[[i]]):
there is no package called 'rlang'
Error : package 'AnnotationDbi' could not be loaded
ERROR: lazy loading failed for package 'org.Mm.eg.db'
* removing 'D:/R-3.5.1/library/org.Mm.eg.db'
In R CMD INSTALLlibrary("ChIPseeker")
library("org.Mm.eg.db")
library("TxDb.Mmusculus.UCSC.mm10.knownGene")
library("clusterProfiler")
txdb <- TxDb.Mmusculus.UCSC.mm10.knownGene2.加载数据
setwd("F:/Data/chip_seq/aligned")
suz12<-readPeakFile("suz12_peaks.narrowPeak")3.查看peak在全基因组的位置
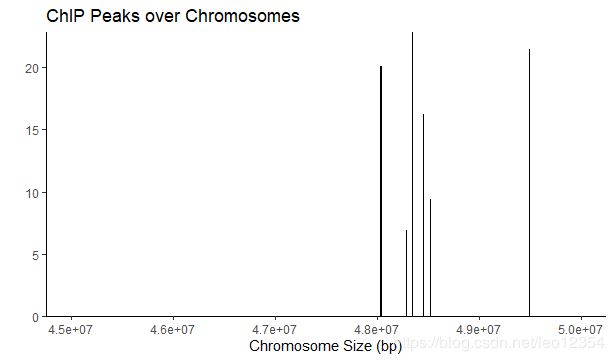
covplot函数可以计算peak 在染色体上的覆盖区域,并可视化。
covplot(suz12,weightCol=5)结果如下,和 ChIP分析流程好像不太一样,可能是index不一样所致。

还可以定义具体的染色体和具体位置
covplot(suz12, weightCol=5, chrs=c("chrX"), xlim=c(4.5e7, 5e7))结果展示:
4.ChIP peaks结合TSS 区域的情况
TSS:transcription start site
首先,计算ChIP peaks结合在TSS区域的情况。这就需要准备TSS区域,这一般定义在TSS位点的侧翼序列(默认-3000~+3000),我这儿改了一下,1500。
然后比对 map到这些区域的peak,并生成tagMatrix。
再做一个热图,Heatmap of ChIP binding to TSS regions
promoter <- getPromoters(TxDb=txdb, upstream=1500, downstream=1500)
tagMatrix <- getTagMatrix(suz12, windows=promoter)
tagHeatmap(tagMatrix, xlim=c(-1500, 1500), color="green")焕发着春天的生机,像草一样出现在了屏幕上。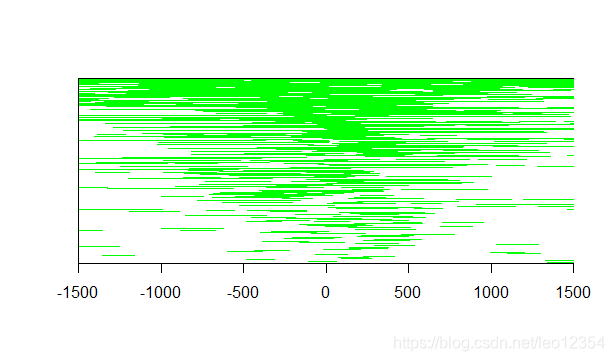
也可以这样一步生成,简单直接。
peakHeatmap(suz12, TxDb=txdb, upstream=2000, downstream=3000, color="blue")>> preparing promoter regions... 2020-05-08 15:53:48
>> preparing tag matrix... 2020-05-08 15:53:48
>> generating figure... 2020-05-08 15:54:27
>> done... 2020-05-08 15:54:39 还是挺快的。

5.Average Profile of ChIP peaks binding to TSS region
Confidence interval estimated by bootstrap method
plotAvgProf(tagMatrix, xlim=c(-1500, 1500),
conf=0.95,resample = 1000,
xlab="Genomic Region (5'->3')", ylab = "Read Count Frequency")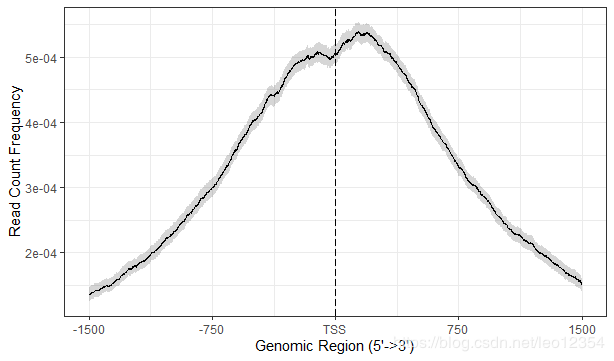
6.peaks注释
annotatePeak函数进行peaks注释,可以定义TSS(转录起始位点)区域,默认情况下TSS定义为-3kb到+ 3kb。
annotatePeak的输出是csAnno格式。 ChIPseeker中国的as.GRanges函数将csAnno转换为GRanges格式,as.data.frame将csAnno转换为data.frame,然后通过write.table将其导出到文件。
TxDb.Hsapiens.UCSC.hg38.knownGene和TxDb.Hsapiens.UCSC.hg19.knownGene分别对应人类基因组hg38和hg19,TxDb.Mmusculus。UCSC.mm10.knownGene和TxDb.Mmusculus.UCSC.mm9.knownGene则对应小鼠基因组mm10和mm9。
用户还可以通过R makeTxDbFromBiomart 和makeTxDbFromUCSC 从 UCSC Genome Bioinformatics和BioMart数据库检索准备自己的TxDb对象。然后进行峰值注释。
所有的峰值信息都会保存在输出文件中。其中包含peak最近的gene的位置和链的信息,从peak到最近的gene的TSS的距离等。鉴于某些信息可能的重叠,ChIPseeker采取以下优先级别:
Promoter
5’ UTR
3’ UTR
Exon
Intron
Downstream
Intergenic
摘自 ChIP分析流程。
结果:
>> preparing features information... 2020-05-08 17:40:02
>> identifying nearest features... 2020-05-08 17:40:04
>> calculating distance from peak to TSS... 2020-05-08 17:40:06
>> assigning genomic annotation... 2020-05-08 17:40:06
>> adding gene annotation... 2020-05-08 17:40:43
'select()' returned 1:many mapping between keys and columns
>> assigning chromosome lengths 2020-05-08 17:40:43
>> done... 2020-05-08 17:40:43 7.可视化基因组注释
为了注释给定peak在基因组的位置特征信息,可使用annotatePeak函数,这些信息在输出信息的“annotation”列,包括peak是否在TSS,外显子,5’UTR,3’UTR,内含子或间隔区。很多研究人员对这些注释非常感兴趣。用户可以自己定义TSS区域。
以下几个图可以自己选择,哪个好看有用选哪个
plotAnnoPie(peakAnno)
plotAnnoBar(peakAnno)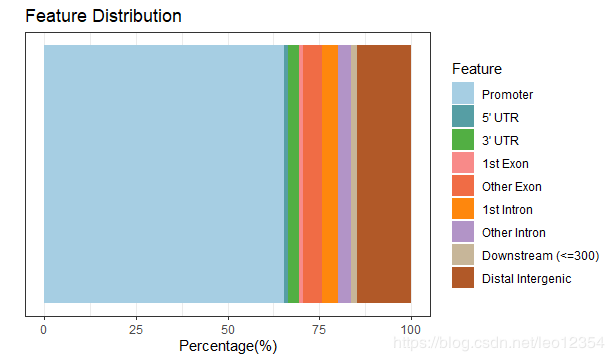
vennpie(peakAnno)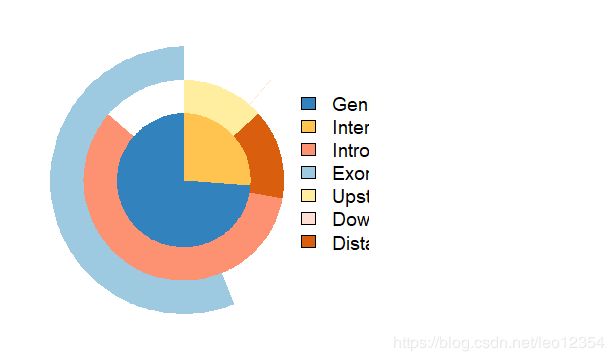
upsetplot(peakAnno)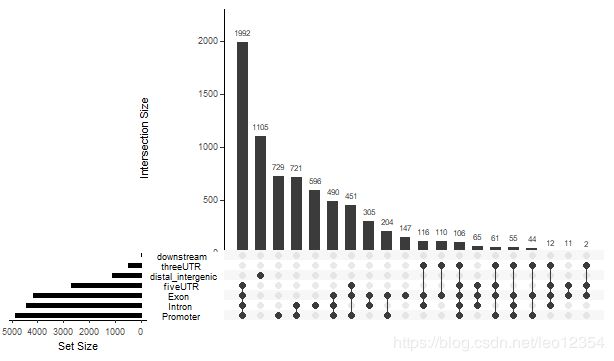
8.Visualize distribution of TF-binding loci relative to TSS
peak(TF结合位点)到最近的gene的TSS之间的距离可以有annotatePeak函数进行计算。作者提供了plotDistToTSS函数计算最近基因的TSS上游和下游的结合位点的百分比,并可视化这种分布。
plotDistToTSS(peakAnno,title="Distribution of transcription factor-binding loci\nrelative to TSS")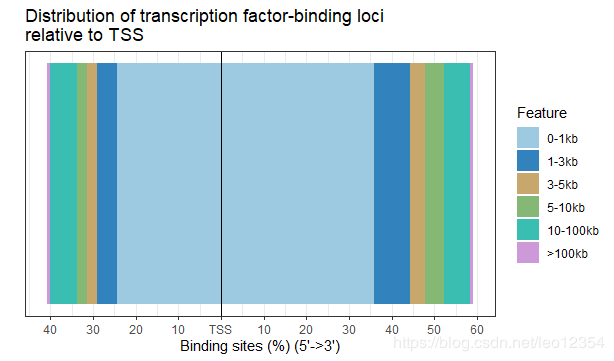
9.多个peak的比较,参考Chip-seq 实战分析流程
多个peak set注释时,先构建list,然后用lapply.list(name1=bed_file1,name2=bed_file2)
RYBP的数据有问题,这里加上去,会一直报错。
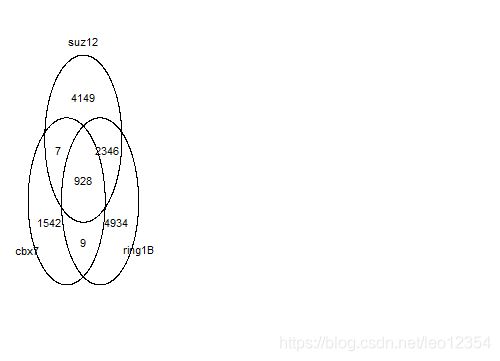
peaks <- list(cbx7=cbx7,ring1B=ring1B,suz12=suz12)
promoter <- getPromoters(TxDb=txdb, upstream=2000, downstream=2000)
tagMatrixList <- lapply(peaks, getTagMatrix, windows=promoter)
plotAvgProf(tagMatrixList, xlim=c(-2000, 2000))
plotAvgProf(tagMatrixList, xlim=c(-2000, 2000), conf=0.95,resample=500, facet="row")
tagHeatmap(tagMatrixList, xlim=c(-2000, 2000), color=NULL)
genes= lapply(peakAnnoList, function(i) as.data.frame(i)$geneId)
peakAnnoList <- lapply(peaks, annotatePeak, TxDb=txdb,tssRegion=c(-2000, 2000), verbose=FALSE)
vennplot(genes)