有人说Y叔的clusterpofiler的GO注释结果和其他工具不一样,后来询问,发现分析物种是拟南芥。这就让我想起了我之前的一篇文章,「Bioconductor」不要轻易相信AnnotationHub的物种注释包, 里面就提到拟南芥的物种包用的注释其实一直都没有更新。究其原因,是因为拟南芥的物种包里的注释一直是从TAIR的FTP下载,而我另一篇文章TAIR周期性更新的注释原来不在FTP服务器上也说了,最新的拟南芥注释信息是要在另外的地方进行下载。
下面就是我拆解Y叔的clusterprofiler验证我的猜想环节。
首先用clusterpofiler::enrichGO调出Y叔的代码是如何写的
enrichGO <- function (gene, OrgDb, keyType = "ENTREZID", ont = "MF", pvalueCutoff = 0.05,
pAdjustMethod = "BH", universe, qvalueCutoff = 0.2, minGSSize = 10,
maxGSSize = 500, readable = FALSE, pool = FALSE)
{
ont %<>% toupper
ont <- match.arg(ont, c("BP", "CC", "MF", "ALL"))
# 这个函数获取GO注释
GO_DATA <- get_GO_data(OrgDb, ont, keyType)
if (missing(universe))
universe <- NULL
if (ont == "ALL" && !pool) {
...
}
从中发现get_GO_data是用于获取基因对应的GO注释,继续深究这个函数
get_GO_data <- function (OrgDb, ont, keytype)
{
...
else {
OrgDb <- load_OrgDb(OrgDb)
kt <- keytypes(OrgDb)
if (!keytype %in% kt) {
stop("keytype is not supported...")
}
# 获取orgDb包中的所有关键字
kk <- keys(OrgDb, keytype = keytype)
# 获取GOALL 和 ONTOLOGYALL
goAnno <- suppressMessages(select(OrgDb, keys = kk, keytype = keytype,
columns = c("GOALL", "ONTOLOGYALL")))
goAnno <- unique(goAnno[!is.na(goAnno$GOALL), ])
assign("goAnno", goAnno, envir = GO_Env)
assign("keytype", keytype, envir = GO_Env)
assign("organism", get_organism(OrgDb), envir = GO_Env)
}
...
}
被我省略的部分基本都是为了判断是否已经缓存了注释信息,或者是返回结果和我们的目的无关,核心的代码是用select获取物种注释包的GOALL和ONTOLOGYALL内容
kk <- keys(org, keytype = "TAIR")
goAnno <- select(org, keys = kk, keytype = "TAIR",
columns = c("GOALL", "ONTOLOGYALL"))
所谓的GOALL指的是一个GO及其它的所有父级词条,所以我们应该用"GO"进行后续搜索。
下一步是找到拟南芥中其中一个基因对应GO。先安装或者加载拟南芥的物种注释包
BiocManager::install("org.At.tair.db")
library("org.At.tair.db")
org <- org.At.tair.db
直接输入org可以看到注释包所用的数据来源
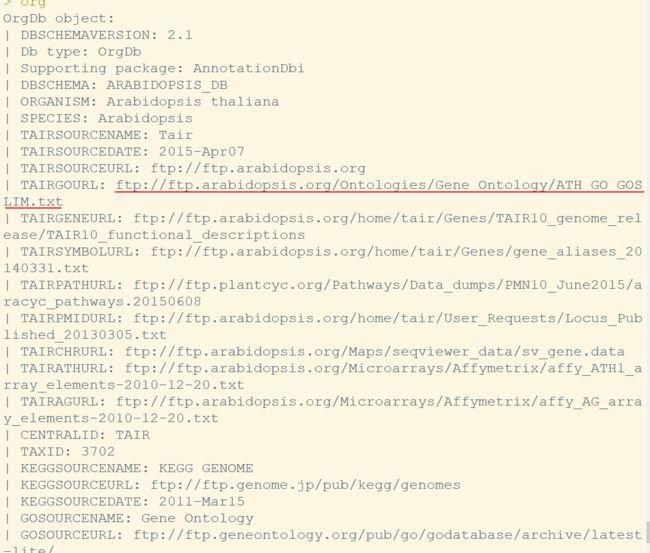
接下来,挑选一个基因输出对应的GO
goAnno <- select(org, keys = kk, keytype = "TAIR",
columns = c("GO"))
goAnno[goAnno$TAIR == "AT1G01010",]
TAIR GO EVIDENCE ONTOLOGY
1 AT1G01010 GO:0003700 ISS MF
2 AT1G01010 GO:0005634 ISM CC
3 AT1G01010 GO:0006888 RCA BP
4 AT1G01010 GO:0007275 ISS BP
5 AT1G01010 GO:0043090 RCA BP
如下是我在"20171231"版ATH_GO_GOSLIM.txt查询该基因的结果,我们发现该基因目前拥有13个GO注释,比原来多了8条。
grep '^AT1G01010' ATH_GO_GOSLIM.txt | cut -f 1,6,8,10
AT1G01010 GO:0005634 C ISM
AT1G01010 GO:0006355 P IEA
AT1G01010 GO:0006355 P ISS
AT1G01010 GO:0006355 P ISS
AT1G01010 GO:0006355 P ISS
AT1G01010 GO:0006355 P IEA
AT1G01010 GO:0005634 C IEA
AT1G01010 GO:0006351 P IEA
AT1G01010 GO:0006355 P IEA
AT1G01010 GO:0003677 F IEA
AT1G01010 GO:0007275 P ISS
AT1G01010 GO:0016021 C IEA
AT1G01010 GO:0003700 F ISS
而在物种包所用的"20150409"版本中,你能找到和org.At.tair.db一模一样的注释内容
grep '^AT1G01010' ATH_GO_GOSLIM.txt | cut -f 1,6,8,10
AT1G01010 GO:0007275 P ISS
AT1G01010 GO:0005634 C ISM
AT1G01010 GO:0003700 F ISS
AT1G01010 GO:0043090 P RCA
AT1G01010 GO:0006888 P RCA
所以clusterprofiler没有问题,而是Bioconductor提供的注释包不准确,那我们就应该跳过中间商,直接作分析。
根据「[clusterProfiler] buildGOmap」一文中提到的:
..如果你分析的是GO,并且你的注释只有直接注释,那么你可以用
buildGOmap把间接注释加上去,然后再用enricher/GSEA分析,所以现在的流程是buildGOmap(optional and only for GO) -> enricher/GSEA。在现在clusterProfiler里,buildGOmap不会产生文件,而是输出一个data.frame,做为background annotation输入给enricher(超几何检验)或GSEA。
那么正确的做法是,先提取出ATH_GO_GOSLIM中所需的列
cat ATH_GO_GOSLIM.txt | cut -f 1,6,8,10 > ATH_GO_TERM.txt
可以根据http://www.geneontology.org/page/guide-go-evidence-codes,使用grep对GO进行过滤。
然后R语言中导入数据, 为GO的BP, CC, MF 分为构建GOMAP(间接注释)
ATH_GOTERM <- read.table("/data/database/TAIR/20171231/ATH_GO_TERM.txt",
sep="\t")
colnames(ATH_GOTERM) <- c("geneID","GOTerm","Ont","Source")
# BP
ATH_GOTERM_BP <- ATH_GOTERM[ATH_GOTERM$Ont == "P",]
gomap_BP <- data.frame(GO=ATH_GOTERM_BP$GOTerm, gene=ATH_GOTERM_BP$geneID)
gomap_BP <- buildGOmap(gomap_BP)
# MF
ATH_GOTERM_MF <- ATH_GOTERM[ATH_GOTERM$Ont == "F",]
gomap_MF <- data.frame(GO=ATH_GOTERM_MF$GOTerm, gene=ATH_GOTERM_MF$geneID)
gomap_MF <- buildGOmap(gomap_MF)
# CC
ATH_GOTERM_CC <- ATH_GOTERM[ATH_GOTERM$Ont == "C",]
gomap_CC <- data.frame(GO=ATH_GOTERM_CC$GOTerm, gene=ATH_GOTERM_CC$geneID)
gomap_CC <- buildGOmap(gomap_CC)
使用enricher进行富集分析
# DEG_GENE_ID 指的是你实际的差异表达分析得到基因名
library(GO.db)
goname_BP <- AnnotationDbi::select(x=GO.db, keys = gomap_BP$GO, keytype = "GOID",columns = "TERM" )
# enricher
ego <- enricher(DEG_GENE_ID,TERM2GENE = gomap, TERM2NAME=goname_BP)
Y叔的clusterProfielr还有一个神奇的工具叫做simplify能够去除冗余的GO,但是目前只认enrichGO的结果。怎么办呢?Y叔提供了一个解决方案:
ego@ontology <- "BP" # 按照需求改成,CC或MF
simplify(ego)
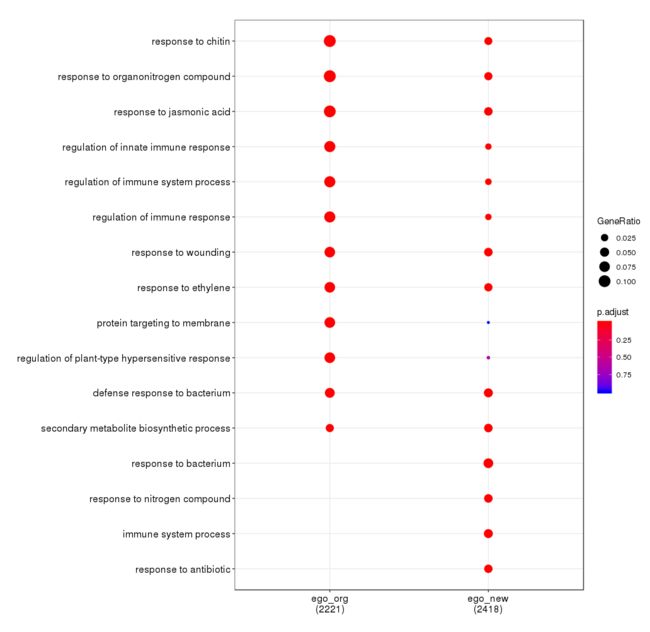
最后以我自己一个分析结果为例,展示下以拟南芥org包作为输入的ecnrichgo和跳过org包用ericher之间的区别。
dotplot(merge_result(list(ego_org= ego_org@resul, ego_new=ego_new@result)))
参考资料
[clusterProfiler] buildGOmap
GO富集分析中的间接注释
SCIENCE文章用了DAVID被吐槽
「Bioconductor」不要轻易相信AnnotationHub的物种注释包
TAIR周期性更新的注释原来不在FTP服务器上