本文首发于我的个人博客, http://xuzhougeng.top/
往期回顾:
- 使用ArchR分析单细胞ATAC-seq数据(第一章)
- 使用ArchR分析单细胞ATAC-seq数据(第二章)
- 使用ArchR分析单细胞ATAC-seq数据(第三章)
- 使用ArchR分析单细胞ATAC-seq数据(第四章)
- 使用ArchR分析单细胞ATAC-seq数据(第五章)
- 使用ArchR分析单细胞ATAC-seq数据(第六章)
- 使用ArchR分析单细胞ATAC-seq数据(第七章)
- 使用ArchR分析单细胞ATAC-seq数据(第八章)
- 使用ArchR分析单细胞ATAC-seq数据(第九章)
- 使用ArchR分析单细胞ATAC-seq数据(第十章)
- 使用ArchR分析单细胞ATAC-seq数据(第十一章)
- 使用ArchR分析单细胞ATAC-seq数据(第十二章)
- 使用ArchR分析单细胞ATAC-seq数据(第十三章)
- 使用ArchR分析单细胞ATAC-seq数据(第十四章)
- 使用ArchR分析单细胞ATAC-seq数据(第十五章)
第16章 使用ArchR进行轨迹分析
ArchR在ArchRProject中低维子空间对细胞进行排序来创建细胞轨迹,实现伪时间中细胞排序。我们之前已经在二维UMAP子空间中执行了这种排序,但是ArchR改进了这种方法,使其能够在N维子空间(即LSI)内进行对齐。
- 首先,ArchR需要用户提供一个轨迹骨架描述细胞分组/聚类的大致分化方向。例如,用户提供的细胞编号为HSC, GMP, Monocyte,这表示细胞以干细胞作为起点,然后是祖细胞,最后是分化细胞。这种分化方向信息来自于已有的生物学相关的细胞轨迹发育的背景知识。
- 其次,ArchR计算每个聚类在N维空间的平均坐标,对于聚类里的每个细胞,计算其相对于平均指标的欧几里得距离,最后只保留前5%的细胞。
- 然后,ArchR根据轨迹计算cluster i里的每个细胞和cluster i+1平均坐标的距离,根据距离计算拟时间向量。之后依次遍历所有的cluster执行运算。根据每个细胞相对于细胞分组/聚类平均坐标的欧几里得距离,ArchR能够为每个细胞确定N维坐标和它的拟时间值从而成为轨迹的一部分。
- 然后,ArchR使用
smooth.spline函数根据拟时间值讲连续的轨迹拟合到N维坐标中。 - 接着,ArchR根据欧氏距离最近细胞沿着流形依次将所有细胞对齐到轨迹上。
- 最后,ArchR将对其后的值缩放到100,并将其保存在
ArchRProject中用于下游分析。
ArchR可以根据Arrow文件里的特征创建拟时间趋势矩阵。例如,ArchR可以分析随着拟时间发生变化的TF deviation, 基因得分,整合基因表达量,从而鉴定随着细胞轨迹动态变化的调节因子或调控元素。
- 首先,ARchR将细胞沿着细胞轨迹进行分组,组数由用户定义(默认是1/100)
- 然后,ArchR使用用户定义平滑滑窗(默认是9/100)对矩阵的特征进行平滑处理(调用
data.table::frollmean函数) - 最后,ArchR会返回一个
SummarizedExperiment对象,它是一个平滑后的拟时间 X 特征矩阵,用于下游分析。
ArchR还可以根据以下信息对任意两个平滑后拟时间 X 特征矩阵进行关联分析,例如匹配的命名(如chromVAR TF deviations的正向调控因子和基因得分/整合谱),之前章节提到的利用低重叠细胞聚集的基因组位置重叠方法(例如 peak-to-gene 关联)。最终,ArchR能为细胞轨迹的整合分析提供帮助,解释多组学之间动态调控关系。
16.1 髓系轨迹-单核细胞分化
这一节,我们会创建一个细胞估计用以模拟HSC发育成分化后单核细胞的过程。 在开始之前,我们先来检查保存在cellColData中的聚类和之前定义的细胞类型,分别是"Clusters"和"Clusters2"。我们通过UMAP来进行展示,从中找到我们感兴趣的细胞类型。
p1 <- plotEmbedding(ArchRProj = projHeme5, colorBy = "cellColData", name = "Clusters", embedding = "UMAP")
p2 <- plotEmbedding(ArchRProj = projHeme5, colorBy = "cellColData", name = "Clusters2", embedding = "UMAP")
ggAlignPlots(p1, p2, type = "h")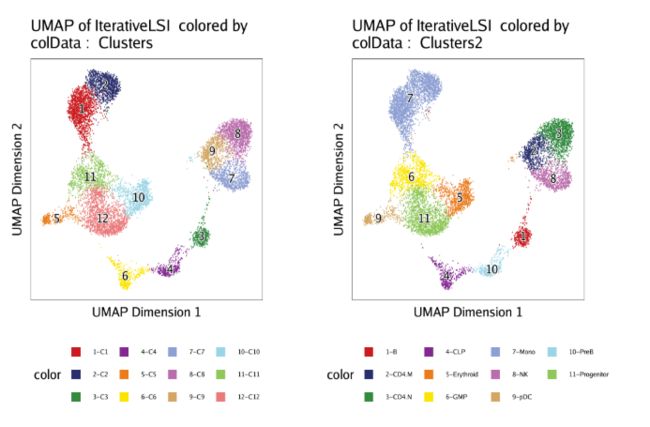
16.1.1 拟时间UMAP和特征单独作图
我们接下来会用到"Clusters2"里面定义的细胞类型。和之前提到的那样,我们会以干细胞("Progenitor")-骨髓祖细胞("GMP")-单核细胞("Mono")这个分化过程创建轨迹。
第一步,我们需要以用一个向量记录轨迹骨干,里面按顺序记录发育各过程中细胞分组的标签。
trajectory <- c("Progenitor", "GMP", "Mono")第二步,我们使用addTrajectory()创建轨迹,并加入到ArchRProject中。我们将其命名为"MyeloidU"。该函数的作用就是在cellColData中新建一列,命名为"MyeloidU",然后记录每个细胞在轨迹中拟时间值。不在轨迹中的细胞记做NA
projHeme5 <- addTrajectory(
ArchRProj = projHeme5,
name = "MyeloidU",
groupBy = "Clusters2",
trajectory = trajectory,
embedding = "UMAP",
force = TRUE
)我们可以检查下该信息,你会发现每个细胞都有一个唯一值,值的取值范围是0-100。我们将NA的细胞过滤掉,因为它们不属于轨迹。
head(projHeme5$MyeloidU[!is.na(projHeme5$MyeloidU)])
# [1] 46.07479 53.75027 44.82834 43.18828 47.49617 43.21015我们使用plotTrajectory()函数绘制轨迹,它会用UMAP进行可视化,用拟时间值进行上色,箭头标识轨迹的发育方向。非轨迹的细胞会以灰色标识。在这个例子中,我们使用colorBy = "cellColData"和name="MyeloidU"让ArchR使用cellColData里"MyeloidU"作为拟时间轨迹。参数中trajectory和name都是"MyeloidU",可能不太容易理解,ArchR根据trajectory来提取细胞,根据name来对细胞进行着色。
p <- plotTrajectory(projHeme5, trajectory = "MyeloidU", colorBy = "cellColData", name = "MyeloidU")
p[[1]]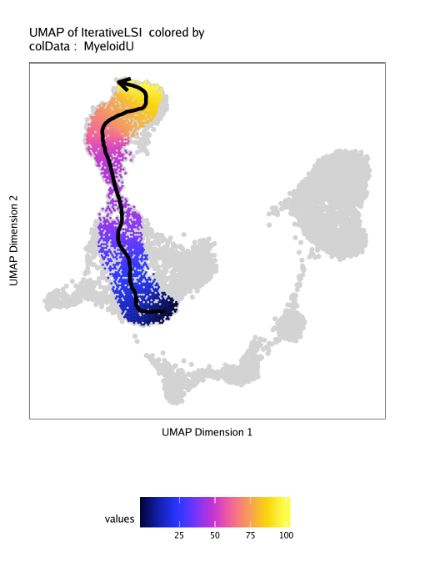
使用plotPDF()保存为可编辑的矢量图
plotPDF(p, name = "Plot-MyeloidU-Traj-UMAP.pdf", ArchRProj = projHeme5, addDOC = FALSE, width = 5, height = 5)我们还可以在UMAP嵌入图中展示轨迹和其他特征。这可以用来展示和我们轨迹有关的特征。
如果你还没有在projHeme5项目中加入推测权重,请先运行下面的代码
projHeme5 <- addImputeWeights(projHeme5)然后我们绘制"MyeloidU"轨迹,只不过颜色来自于"CEBPB"的基因得分,这是一个已知的调控单核细胞功能的基因,会随着发育而活跃。我们通过colorBy参数来指定表达量矩阵,使用name来指定特征
p1 <- plotTrajectory(projHeme5, trajectory = "MyeloidU", colorBy = "GeneScoreMatrix", name = "CEBPB", continuousSet = "horizonExtra")重复上面的代码,只不过这次使用的是GeneIntegrationMatrix作为基因表达量矩阵
p2 <- plotTrajectory(projHeme5, trajectory = "MyeloidU", colorBy = "GeneIntegrationMatrix", name = "CEBPB", continuousSet = "blueYellow")plotTrajectory函数实际上返回了一系列图。列表中第一个图是UMAP嵌入图,根据函数调用进行上色。
通过比较基因得分和基因表达量的UMAP图,不难发现CEBPB基因在单核细胞中特异性表达,出现在你时序轨迹的后半部分。
ggAlignPlots(p1[[1]], p2[[1]], type = "h")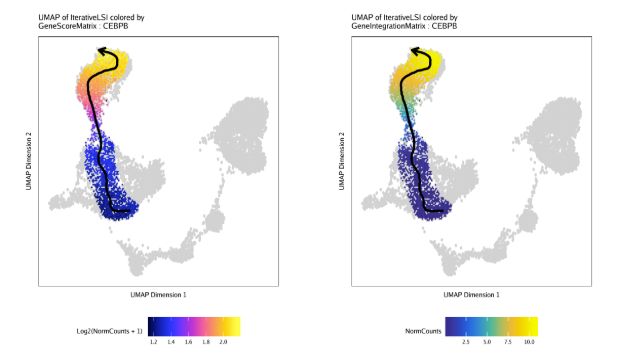
plotTrajectory()函数返回的是一个点图,x轴是拟时间,y轴是对应特征的值,此处是CEBPB的基因得分或基因表达量。下图中的细胞以拟时间进行着色
ggAlignPlots(p1[[2]], p2[[2]], type = "h")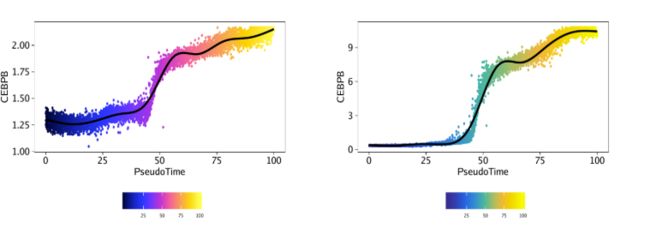
16.1.2 拟时间热图
我们可以用热图的形式对特征随着拟时间变化进行可视化。首先我们用getTrajectory()函数从ArchRProject中提取感兴趣的轨迹,它会返回一个SummarizedExperiment对象。通过设置useMatrix参数,我们可以用motif,基因得分,基因表达量,peak开放性来创建拟时间热图
trajMM <- getTrajectory(ArchRProj = projHeme5, name = "MyeloidU", useMatrix = "MotifMatrix", log2Norm = FALSE)接着将SummarizedExperiment传给plotTrajectoryHeatmap()函数
p1 <- plotTrajectoryHeatmap(trajMM, pal = paletteContinuous(set = "solarExtra"))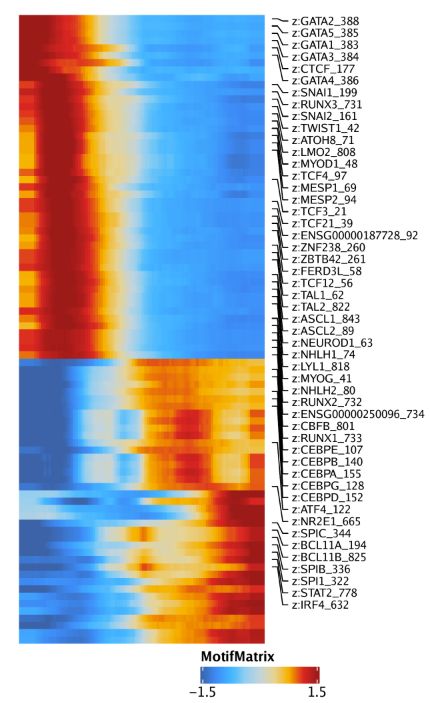
同样的分析可以使用基因得分绘制拟时间热图,只需要设置useMatrix = "GeneScoreMatrix"
trajGSM <- getTrajectory(ArchRProj = projHeme5, name = "MyeloidU", useMatrix = "GeneScoreMatrix", log2Norm = TRUE)
p2 <- trajectoryHeatmap(trajGSM, pal = paletteContinuous(set = "horizonExtra"))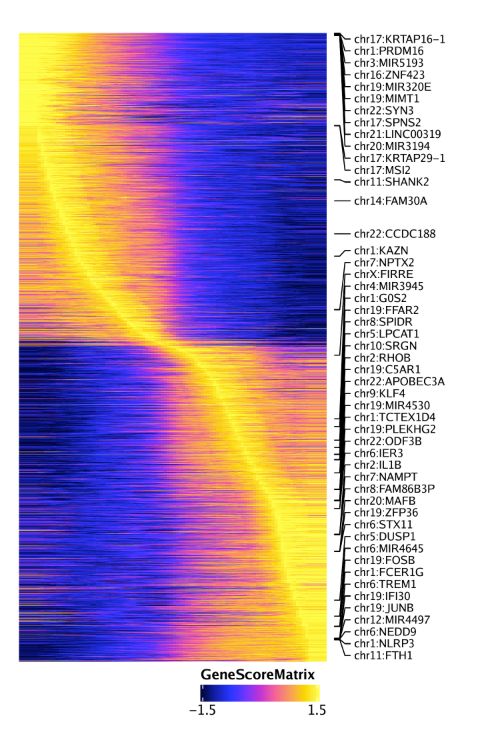
同样的,还可以绘制基因表达量的拟时间热图,设置useMatrix = "GeneIntegrationMatrix".
trajGIM <- getTrajectory(ArchRProj = projHeme5, name = "MyeloidU", useMatrix = "GeneIntegrationMatrix", log2Norm = FALSE)
p3 <- plotTrajectoryHeatmap(trajGIM, pal = paletteContinuous(set = "blueYellow"))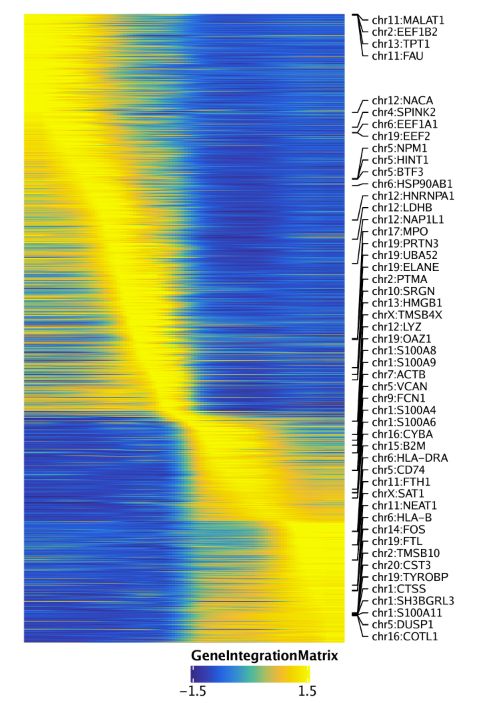
最后,我们可以通过设置useMatrix = "PeakMatrix"绘制peak开放性的拟时间热图
trajPM <- getTrajectory(ArchRProj = projHeme5, name = "MyeloidU", useMatrix = "PeakMatrix", log2Norm = TRUE)
p4 <- plotTrajectoryHeatmap(trajPM, pal = paletteContinuous(set = "solarExtra"))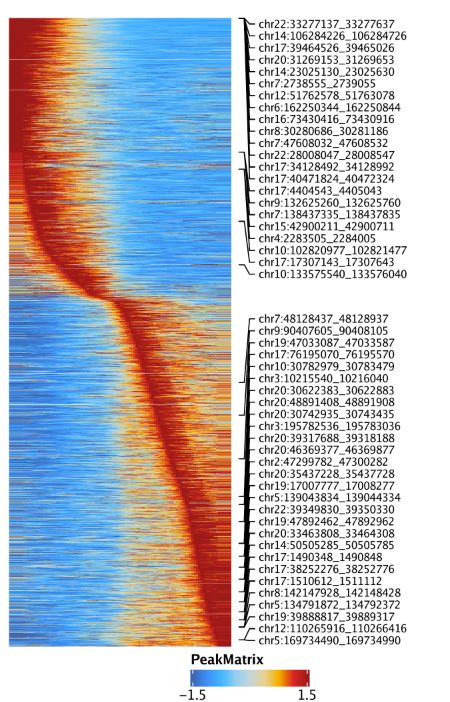
使用plotPDF()保存可编辑的矢量图
plotPDF(p1, p2, p3, p4, name = "Plot-MyeloidU-Traj-Heatmaps.pdf", ArchRProj = projHeme5, addDOC = FALSE, width = 6, height = 8)16.1.3 整合拟时间分析
我们也可以进行整合分析,例如整合基因得分/基因表达量和motif开发状态,鉴定随拟时间变化的正向TF调控因子。这是一个非常有效的手段,例如鉴定分化相关的驱动因子。我们可以用correlateTrajectories()函数进行该分析,它接受两个SummarizedExperiment对象,该对象可以用getTrajectories()函数获取。
首先,让我们找到开放状态随拟时间和TF基因的基因得分相关的motif
corGSM_MM <- correlateTrajectories(trajGSM, trajMM)correlateTrajectories()函数的主要输出是一个列表,第一个是DataFrame对象。DataFrame的列名分别是idx1, matchname1, name1和VarAssay1, 对应索引,匹配名,原名,传递给correlateTrajectories()函数的第一个轨迹的特征的方差分位数(variance quantile)。方差分位数是给定特征的标准化值,能用于比较不同来源assay里的相关值。该DataFrame包括所有符合correlateTrajectories()函数里阈值的特征。
corGSM_MM[[1]]
## DataFrame with 36 rows and 12 columns
## idx1 idx2 matchname1 matchname2 name1 name2
##
## 1 82 1081 PRDM16 PRDM16 chr1:PRDM16 z:PRDM16_211
## … … … … … … …
## 36 22097 1565 RXRA RXRA chr9:RXRA z:RXRA_695
## Correlation VarAssay1 VarAssay2 TStat
##
## 1 0.859588836579798 0.999783802481948 0.860344827586207 16.6530781620713
## … … … … …
## 36 0.59509316489084 0.88640982401522 0.890229885057471 7.33039570155334
## Pval FDR
##
## 1 2.47466579579935e-30 4.04855324192774e-27
## … … …
## 36 6.60626612679289e-11 1.25672690505037e-09我们然后从SummarizedExperiment提取相应的轨迹,
trajGSM2 <- trajGSM[corGSM_MM[[1]]$name1, ]
trajMM2 <- trajMM[corGSM_MM[[1]]$name2, ]为了更好的对这些特征排序,我们创建新的轨迹,它的值来自于原先的轨迹的相乘。这可以让我们的热图根据行进行排序
trajCombined <- trajGSM2
assay(trajCombined) <- t(apply(assay(trajGSM2), 1, scale)) + t(apply(assay(trajMM2), 1, scale))我们可以从plotTrajectoryHeatmap()函数返回的结果提取最优的行序。
combinedMat <- plotTrajectoryHeatmap(trajCombined, returnMat = TRUE, varCutOff = 0)
rowOrder <- match(rownames(combinedMat), rownames(trajGSM2))有了这个之后,我们就可以创建配对热图。
首先,我们根据基因得分轨迹创建热图。在rowOrder参数中设置给定的行序
ht1 <- plotTrajectoryHeatmap(trajGSM2, pal = paletteContinuous(set = "horizonExtra"), varCutOff = 0, rowOrder = rowOrder)然后,我们根据motif轨迹创建热图。在rowOrder参数中设置给定的行序
ht2 <- plotTrajectoryHeatmap(trajMM2, pal = paletteContinuous(set = "solarExtra"), varCutOff = 0, rowOrder = rowOrder)通过将两个热图并排排列,我们可以看到两个热图的行是匹配的。你可能会发现该分析同时捕获了GATA3和GATA3-AS1(一个GATA3的反义转录本)。这是由于特征匹配所导致的,我们需要在后续分析时进行手动过滤。
ht1 + ht2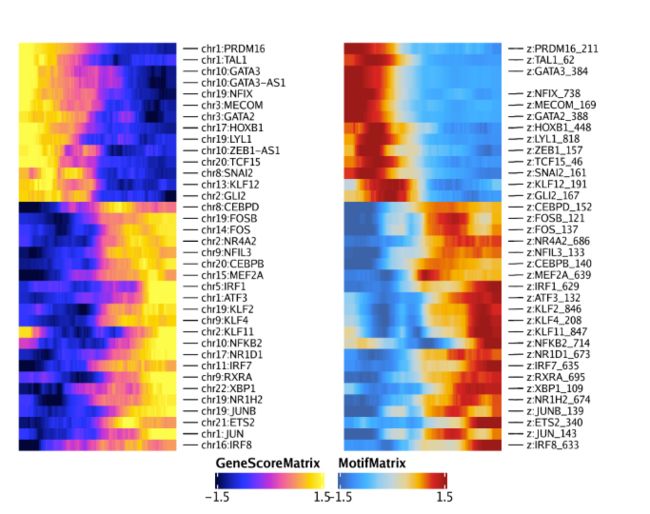
我们可以重复以上的分析,但这次使用GeneIntegrationMatrix而非基因得分。因为这是相同的分析流程,因此代码就不再过多解释
corGIM_MM <- correlateTrajectories(trajGIM, trajMM)
trajGIM2 <- trajGIM[corGIM_MM[[1]]$name1, ]
trajMM2 <- trajMM[corGIM_MM[[1]]$name2, ]
trajCombined <- trajGIM2
assay(trajCombined) <- t(apply(assay(trajGIM2), 1, scale)) + t(apply(assay(trajMM2), 1, scale))
combinedMat <- plotTrajectoryHeatmap(trajCombined, returnMat = TRUE, varCutOff = 0)
rowOrder <- match(rownames(combinedMat), rownames(trajGIM2))
ht1 <- plotTrajectoryHeatmap(trajGIM2, pal = paletteContinuous(set = "blueYellow"), varCutOff = 0, rowOrder = rowOrder)
ht2 <- plotTrajectoryHeatmap(trajMM2, pal = paletteContinuous(set = "solarExtra"), varCutOff = 0, rowOrder = rowOrder)
ht1 + ht2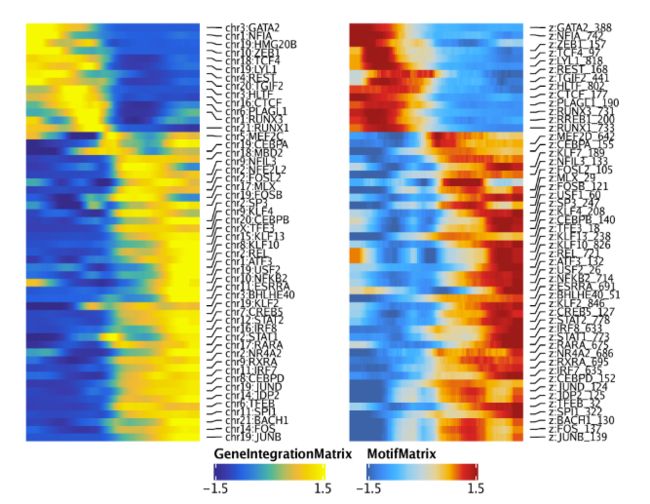
16.2 淋巴轨迹-B细胞分化
第二个例子会根据祖细胞-淋巴祖细胞-前B细胞-分化后B细胞来构建B细胞的分化轨迹。由于该分析从本质上是重复之前单核细胞轨迹分析,因此这里只介绍关键性的代码不同。
第一处是trajectory变量的赋值。
trajectory <- c("Progenitor", "CLP", "PreB", "B")第二处是使用addTrajectory()创建轨迹,参数name=LymphoidU。
projHeme5 <- addTrajectory(
ArchRProj = projHeme5,
name = "LymphoidU",
groupBy = "Clusters2",
trajectory = trajectory,
embedding = "UMAP",
force = TRUE
)第三处, 我们需要注意plotTrajectory里的参数trajectory和name需要改成"LymphoidU"
p <- plotTrajectory(projHeme5, trajectory = "LymphoidU", colorBy = "cellColData", name = "LymphoidU")后续的代码基本上可以照搬上一节。