Protocol
1. hPSC Culture in the Chemically Defined and Feeder-free Condition
-
Adapt the hPSC culture on iMEF feeders to the feeder-free condition. In cases where frozen cells do not survive well when directly recovered in the feeder-free condition, recover cells in iMEF condition first and then adapt to the feeder-free condition.
NOTE: In general, it takes 2 passages for hPSCs cultured on iMEF feeders to be adapted to the feeder-free condition.
Change the medium every day and passage the hPSCs when the cells have reached ~80% confluency. In general, passage hPSCs at ~ 1:6 - 1:15 ratios every 4 - 6 days. Add 10 μM ROCK inhibitor Y-27632 when thawing or passaging the cells.
Prior to seeding the hPSCs, pre-coat culture dishes with 5 μg/mL (1 mL/10 cm2) truncated recombinant human form of vitronectin (VTN) for at least 1 h at room temperature (RT). Also, prepare complete chemically defined medium by adding supplement into the basal medium.
Remove the culture medium, wash the cells once with PBS without Ca2+ and Mg2+, and treat the cells with 0.5 mM EDTA for ~ 2 - 5 min at RT.
Collect the dissociated hPSCs and spin down the cells at 200 x g for 5 min. Resuspend the pelleted hPSCs in the complete medium and seed the cells on VTN-coated plates.
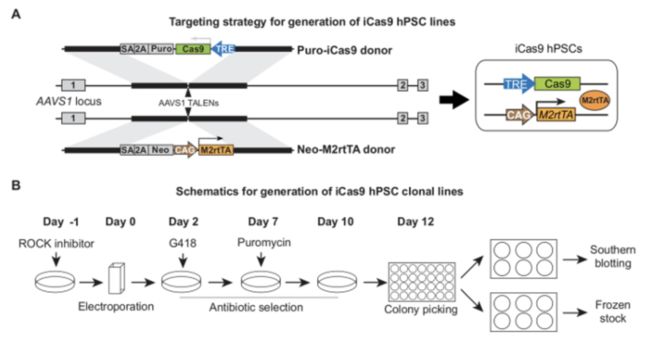
2.Generation of iCas9 hPSC Lines
-
Order and amplify the following plasmids:
AAVS1-TALEN-L, AAVS1-TALEN-R, AAVS1-Neo-M2rtTA, and AAVS1-Puro-iCas9.
NOTE: To avoid unexpected recombination events, use recombination-deficient **Stbl3 **competent cells for the transformation and amplification of plasmids at 30 °C.
Typically, prepare the hPSCs in one 10-cm dish (~ 1 x 107 cells if ~ 80% confluent) for one targeting experiment.
**NOTE: **Since electroporation usually causes significant cell death and TALEN-mediated gene targeting in the AAVS1 locus requires antibiotic selection, a relatively large number of cells need to be seeded to identify correctly targeted cells. Optimization for the drug concentrations is recommended for each cell line and each culture condition.On Day -1, (the day before electroporation), add 10 μM ROCK inhibitor during the media change.
On Day 0, (the day of electroporation), prepare VTN-coated plates in advance.
Dissociate the hPSCs into single cells using 1x dissociation reagent. Briefly, remove the culture medium, wash the cells once with PBS without Ca2+ and Mg2+, and treat the cells with 1x dissociation reagent at 37 °C for ~ 3 min. Aspirate the dissociation reagent before the cells have detached. With gentle pipetting, disperse the hPSCs into a single-cell suspension in 10.5 mL of complete medium.
Take 0.5-mL cell suspension to count the cell number using an automated cell counter. Pellet the hPSCs at 200 x g for 5 min and resuspend the cells in cold (4 °C) PBS at 12.5 x 106 cells/mL.
Add the plasmids into 800-μL hPSC suspension (12.5 x 106 cells/mL) and mix well. Transfer the mixture to a 0.4-cm electroporation cuvette and keep on ice for ~ 5 min.
Electroporate the cells using an electroporation system at 250 V and 500 μF; the time constant observed after electroporation is typically 9 - 13 ms.
-
After electroporation, transfer the cells to a 15-mL conical tube with 5 mL of pre-warmed complete medium.
Critical for successful targeting: Use healthy proliferating hPSCs and handle the cells very gently when transferring, resuspending, and plating the cells after electroporation.
Pellet the cells at 200 x g for 5 min. Resuspend the cells in 10 mL of complete medium with 10 μM ROCK inhibitor and plate 1, 2.5, and 5 x 106 cells onto each of the three VTN-coated, 10-cm dishes; this ensures that at least one of the plates will have sufficient colonies at single-cell clonal density for colony picking.
On Day 1 (the day after electroporation), change the medium.
On Days 2 - 5, start neomycin selection when the cells are ~ 60% confluent. Change the medium daily with 500 μg/mL G418 sulfate;
significant cell death due to selection is typically observed 2 days after G418 selection.On Day 6, change the medium without antibiotic selection.
On Days 7-9, start puromycin selection. Change the medium daily with 1 μg/mL puromycin dihydrochloride; significant cell death should be observed the next day.
-
On Day 10, start changing the medium daily without antibiotic selection until the hPSC single-cell colonies reach 1 - 2 mm in diameter.
NOTE: Typically, 50 colonies in a 10-cm dish are observed with 2.5 x 106 hPSCs plated on Day 0.
Pick 12 - 24 colonies under a stereomicroscope. Mechanically disaggregate the hPSC colonies into small pieces (~ 10 pieces per colony)
using a 23-G needle (a 200-μL pipet tip is also fine) and transfer the cells directly into VTN-coated 24-well plates.Change the medium daily until the cells become confluent. Passage the cells in each well of the 24-well plates into duplicate wells of 6-well
plates.When the cells become confluent in the 6-well plates, use one well for a frozen stock and the other well for genomic DNA extraction for
further characterization.-
Characterize and validate the established iCas9 lines by PCR genotyping, Southern blotting, RT-qPCR analysis, karyotyping, and
pluripotency assay.
 Generation_of_iCas9_hPSC_Lines.jpg
Generation_of_iCas9_hPSC_Lines.jpg
3. Generation of hPSC Mutant Lines Using the iCRISPR System
1. Generation of hPSC knockout lines
1. gRNA design and production
Choose target regions in the gene of interest to maximize the possibility of disrupting wild-type protein function. For well-annotated genes, choose a target region upstream of an essential functional domain. Alternatively, design gRNAs to target a region downstream of the start codon. Choose at least 2 different regions for a gene of interest.
-
Design gRNAs using the online CRISPR design tool http://crispr.mit.edu. For each target region, design 3 gRNAs with low potential off-targets and use the one with the highest targeting efficiency for generating clonal mutant line.
NOTE: In order to achieve high genome editing efficiency, it is recommended to deliver gRNA as RNA oligos instead of as plasmid DNA because of the higher transfection efficiency of small RNAs compared to plasmids in past experience.
Order 120 nucleotide (nt) DNA oligos containing the T7 promoter sequence, the variable 20-nt crRNA recognition sequence (N)20 (do not include the PAM sequence), and the constant chimeric guide sequence. Dilute the oligos to 100-μM stock solution in ddH2O and prepare 250-nM as working solution.
NOTE: TAATACGACTCACTATAGGG (N)20GTTTTAGAGCTAGAAATAGCAAGTTAAAATAAGGCTAGTCCGTTATCAACTTGAAAAAGTGGCACCGAGTCGGTGCTTTTPCR-amplify the oligos using the T7F and TracrR primers to produce the double-stranded DNA (dsDNA) template for gRNA in vitro transcription (IVT). Use 50 μL of PCR reaction mixture and PCR cycle conditions.
Use a high-yield T7 transcription kit for in vitro gRNA transcription with the PCR-amplified template in 20 μL, in vitro gRNA transcription mix as per the manufacturer's instructions. Purify the gRNA products using the transcription clean-up kit as per the manufacturer's instructions.
Elute gRNAs following the high-throughput purification protocol as per manufacturer's instructions (typically ~ 50 - 100 μg) in 100 μL of elution buffer. Adjust the concentration to 320 ng/μL (10 μM) when possible and store at -80 °C until use.
2. PCR and Sanger sequencing primer design
- Design and validate PCR primers amplifying the target region, with product sizes typically ranging from ~ 500 - 1,000 bp.
- Design Sanger sequencing primers binding internally to the PCR products to allow direct sequencing of the PCR products without purification.
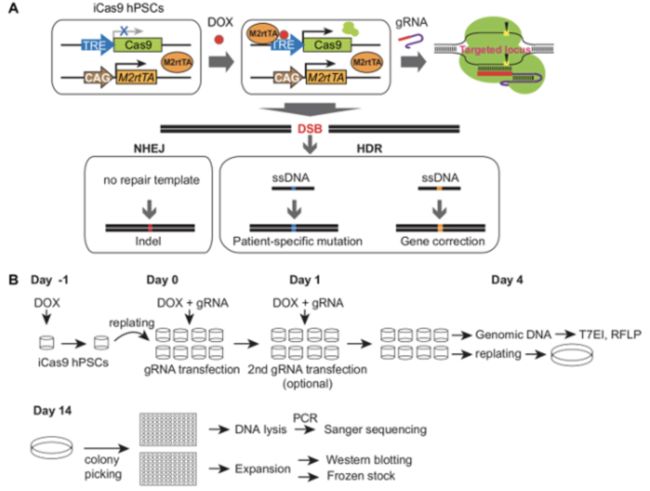
3. gRNA transfection in iCas9 hPSCs
On Day -1, treat iCas9 cells with 2 μg/mL doxycycline 24 h before gRNA transfection.
On Day 0 of gRNA transfection, prepare VTN-coated plates in advance.
Dissociate iCas9 cells into single cells using 1x dissociation reagent, as described in step 2.5.
Pellet the hPSCs at 200 x g for 5 min and resuspend the cells at ~ 0.5 x 106 cells/mL in complete medium supplemented with 2 μg/mL doxycycline and 10 μM ROCK inhibitor.
Plate 0.5 mL of the resuspended cells into individual wells of 24-well plates. Prepare additional wells to serve as non-transfected controls.
-
For each gRNA, make the following transfection mixtures:
Mix A: 50 μL of reduced serum medium + 1 μL of gRNA (10 μM);
Mix B: 50 μL of reduced serum medium + 3 μL of transfection reagent.
Combine Mix A and B to make 100 μL mixture. Incubate for 5 min at RT. Add 50 μL of the mixture to cells in the duplicate wells of the 24-well plates and mix well.
On Day 1, perform a second transfection if needed to further increase the targeting efficiency. Otherwise, change the medium without doxycycline.
On Days 2 - 3, change the medium daily.
On Day 4, extract genomic DNA from one well of each transfected and non-transfected control cell using a DNA extraction kit.Adjust the concentration to 50 ng/μL.
PCR-amplify the target regions flanking the gRNA targeting sequences and estimate the editing efficiency using endonuclease I (T7EI) digestion or the restriction fragment length polymorphism (RFLP) assay, as described previously.
4.T7EI assay
PCR-amplify the target region using the primers designed and validated in step 3.1.
-
Prepare the mixtures (see Table 6) and perform DNA denaturation and hybridization of the PCR products using conditions
outlined in Table 7.NOTE: Based on our experience, it is generally not necessary to purify PCR products for the T7EI assay when using our PCR condition. However, purification could be beneficial in other conditions.
Perform a T7EI digestion at 37 °C for 30 min using 10 μL of denatured and hybridized PCR product and 0.2 μL (2 U) of T7E1 (10 U/μL).
-
Resolve the T7E1-digested PCR samples by gel electrophoresis. Use ImageJ to determine the relative band intensities of cut and uncut DNA.
Calculate the indel frequency using the formula: (1 - (√(1-(b+c))/(a+b+c))) x 100, where a is the intensity of the undigested PCR product and b and c are the intensities of the T7E1-cleaved products.
5. RFLP assay
NOTE: In cases where a restriction site is in close proximity (< 5 bp) to a Cas9 cleavage site (3 bp 5' of the PAM sequence), an RFLP assay can be performed to quantify the indel frequency.
Use the same PCR products as described in step 3.1.4.1.
Digest the PCR product with a restriction enzyme that contains a restriction site in close proximity to the Cas9 cleavage site.
-
Resolve the digested PCR samples by gel electrophoresis. Use ImageJ to determine the relative band intensities of cut and uncut DNA.
Calculate the indel frequency using the formula: a / (a+b+c) x 100, where a is the intensity of the undigested PCR product and b and c are the intensities of the digested products.
6. Establishment of clonal mutant lines
**NOTE: **Genome editing in hPSCs using the iCRISPR system with gRNA transfection is highly efficient, and no antibiotic selection is needed. To establish clonal lines, it is necessary to seed cells at a relatively low density to ensure the formation of single-cell-derived colonies.
- Identify gRNAs with the highest editing efficiency (using the T7EI or RFLP assay) and with good cell survival. Use the corresponding duplicate well for clonal mutant line establishment.
- Dissociate the hPSCs into a single-cell suspension using 1x dissociation reagent, as described in step 2.5. Replate 500, 1,000, and 2,000 cells onto each of the three VTN-coated, 10-cm dishes.
- Change the medium daily until the single-cell colonies reach ~ 2 mm in diameter.
- Pick 24 - 48 colonies for each gRNA, depending on the estimation of targeting efficiency by the T7EI and/or RFLP assay.Mechanically disaggregate each colony into small pieces (~ 10 pieces per colony) using a 23-G needle (a 200-μL pipet tip is also fine) and replate the cells in duplicate VTN-coated, 96-well plates. Use one plate for genomic DNA extraction and Sanger sequencing and the other plate for further expansion.
- When the cells in the 96-well plates have become confluent, extract the genomic DNA (without phenol/chloroform extraction) using a simple protocol, as outlined below.
- Remove the medium and wash the cells once with PBS without Ca2+ and Mg2+ . Add 50 μL of lysis buffer (5 μL of Proteinase K (10 mg/mL), 5 μL of PCR buffer 10x, and 40 μL of ddH2O) to each well of a 96-well plate. Seal the plate using an adhesive film and incubate overnight at 55 °C.
- The next day, transfer the cell lysates into a 96-well PCR plate and incubate for 10 min at 99 °C in a thermocycler to inactivate the Proteinase K.
- PCR-amplify the target region using the same primers as for the T7EI or RFLP assay, using 1 μL of cell lysate as a template.
- Use 1 μL of the PCR product for Sanger sequencing with a primer binding internally to the PCR product.
- Amplify the clones with frameshift indel mutations for frozen stocks. Also, amplify a couple of wild-type clones from the same targeting experiment to serve as isogenic control lines for further experiments.
2. Generation of mutant lines with precise nucleotide alterations
NOTE: Compared with knockout mutants generated by non-homologous end joining (NHEJ), precise nucleotide alteration can be achieved through homology-directed repair (HDR) in the presence of DNA repair templates. Such precise nucleotide alterations allow for the generation of patient-specific mutations in wild-type hPSCs and for the correction of mutations in patient-derived iPSCs.
1. Design of ssDNA as an HDR template
- Design and produce 2 - 3 gRNAs in close proximity to a patient-specific mutation, as described in step 3.1.1.
- Design a single-stranded DNA (ssDNA) containing the patient-specific mutation flanked by ~ 40 - 80 nt of homology on each side as an HDR template.
- To reduce additional cutting after the correct repair, introduce a silent mutation to the ssDNA template in the region within the gRNA recognition sequence and in close proximity to the PAM sequence or in the PAM sequence itself, if possible.
- If possible, design the silent mutation to introduce a novel restriction digestion site as well so that it can be used to estimate the repair efficiency using the RFLP assay.
2. gRNA/ssDNA co-transfection and the establishment of clonal lines
-
Perform the co-transfection of gRNA/ssDNA into iCas9 cells, as described in step 3.1.3, with transfection mixtures A and B.
For each gRNA and non-transfected control, transfect the cells in duplicate wells of 24-well plates.Mix A: 50 μL of reduced serum medium + 1 μL of gRNA (10 μM) + 2 μL of ssDNA (10 μM).
Mix B: 50 μL of reduced serum medium + 3 μL of transfection
reagent. After transfection, extract genomic DNA from one well of each transfected and non-transfected control cell and estimate the repair efficiency using the T7EI and/or RFLP assay.
Identify the gRNA/ssDNA mixture with the highest repair efficiency and good cell survival. Use the corresponding duplicate well for clonal mutant line establishment.
Pick 48 - 96 colonies, depending on the estimation of the targeting efficiency by the T7EI and/or RFLP assay. In general, the efficiency of HDR-mediated mutation is lower than knockout mutation and thus more colonies need to be picked.
-
Sequence, expand, and validate clonal lines, as described in step 3.1.6.
 Efficient_Genetic_Modification_in_hPSCs_Using_the_iCRISPR_System.jpg
Efficient_Genetic_Modification_in_hPSCs_Using_the_iCRISPR_System.jpg
4. In Vitro hPSC Differentiation into Glucose-responsive Pancreatic β Cells
NOTE: In vitro differentiation of hPSC mutants into disease-relevant cell types provides a platform for disease modeling in a dish. The following protocol focuses on the in vitro differentiation of hPSCs into glucose-responsive pancreatic β cells for pancreatic developmental and diabetic studies.
1. hPSC differentiation into definitive endoderm
- Maintain hPSC mutants and wild-type control lines in the chemically defined and feeder-free condition, as described in step 1. 2. To prepare the hPSCs for differentiation, dissociate the hPSCs using 1x dissociation reagent and disperse them into single-cell suspension in complete medium.
- Pellet the cells at 200 x g for 5 min and re-suspend the cells in complete medium with 10 μM ROCK inhibitor. Count the cell number and seed the cells at ~ 1.4 x 105 cells/cm2 on VTN-coated plates.
- Change the medium 24 h after seeding.
- On Day 0, start the differentiation after 48 h, when the cells have reached ~ 80% confluency.
NOTE: To achieve high pancreatic differentiation efficiency, optimizing the seeding density and level of confluency at 48 h is recommended for each individual line. - Aspirate the hPSC medium and rinse the cells once with PBS without Ca2+ and Mg2+.
- Change the medium to differentiation day 0 (d0) medium.
- On Days 1 - 2, change the differentiation medium daily, according to the recipes in Table 8.
- On Day 3, examine the definitive endoderm markers SOX17, FOXA2, and CXCR4 by immunofluorescent staining and flow cytometry analysis.
2. Definitive endoderm differentiation into pancreatic progenitor
- On Days 3 - 9, continue definitive endoderm differentiation toward the pancreatic lineage by changing the medium daily, according to the recipes in Table 9.
- On Day 7, examine the early pancreatic progenitor (PP1) marker PDX1 by immunofluorescent staining and flow cytometry analysis.
- On Day 10, examine the later pancreatic progenitor (PP2) markers PDX1 and NKX6.1. Meanwhile, prepare to transfer PP2 cells to air-liquid interface for further differentiation into pancreatic endocrine cells.
3. Pancreatic endocrine differentiation in air-liquid interface
- Treat the PP2 cells with 10 μM ROCK inhibitor 4 h before dissociation.
- Remove the medium and rinse the cells once with PBS without Ca2+ and Mg2+.
- Add 2 mL of 1x dissociation reagent to PP2 cells in one 10-cm dish and incubate at 37 °C for 2 - 3 min.
- Aspirate the dissociation reagent before the cells have detached. Add 10 mL of BLAR medium and disperse the PP2 cells into single cells by gently pipetting up and down.
- Collect the single-cell suspension. Count the cell number and pellet at 200 x g for 5 min.
- Resuspend the cell pellet at ~ 0.5 x 105 cells/μL in S5 differentiation medium and spot 5 - 10 μL of cells per spot on a transwell insert filter. Place 10 - 15 spots in one 6-well insert and ~ 100 spots in a 10-cm insert.
- Add S5 medium to the bottom of each transwell insert, ~ 1.5 mL for 6-well inserts and ~ 8 mL for 10-cm inserts.
- Change the medium daily with the recipes in Table 10.
- Examine pancreatic endocrine markers PDX1, NKX6.1 NEUROD1, NKX2.2, INSULIN, and GLUCAGON on day 34 by immunofluorescent staining and flow cytometry analysis.
-
Examine hPSC-derived β-like cell function with a glucose-stimulated insulin secretion (GSIS) assay .
Directed_hPSC_Differentiation_into_Glucose-responsive_Pancreatic_β-like_Cells.jpg
References
Genome Editing and Directed Differentiation of hPSCs for Interrogating Lineage Determinants in Human Pancreatic Development