聚类分析与可视化
聚类分析与可视化
——《The Wiley Handbook of Human Computer Interaction》学习心得
小组成员:吴** / 周** / 蒋**
概述
最近,在交互媒体专题设计这门课的课程安排下,我们小组对《The Wiley Handbook of Human Computer Interaction》这本书的第945页至第966页部分进行了翻译和学习。该部分的章节名为 “ Visual Analytics for Comparing Multiple Clustering Results of Bioinformatics Data”,其主要介绍了一款能够帮助生物学家进行多种聚类结果分析的软件——XCluSim。在接下来的文章中,我们将分享我们在学习该章节后的收获和感悟。
什么是聚类分析
百度百科对“聚类分析”一词是这样解释的:聚类分析是指将物理或抽象对象的集合分组为由类似对象组成的多个类的分析过程。简单地来说,聚类分析就是将一组元数据划分为多个类或簇,同一个簇中的数据具有很大的相似性,而不同簇中的数据具有较大的差异性。聚类与分类是不同的,因为聚类所要求划分的类是未知的,这与机器学习中的无监督学习【1】过程相似。也正因为聚类过程是没有明确方向的,所以不同的聚类方法往往会得到不同的结果,这就要求人们对聚类结果的质量进行量化。目前为止,常用的评价指标包括了Purity(纯度)、Entropy(熵)、Accuracy(准确率)、NMI(归一化互信息)、ARI(调整兰德指数)【2】等。
XCluSim的诞生
自从Eisen实验室推广了微阵列数据的聚类分析和可视化【3】,聚类分析已经广泛应用于生物信息学界。随着遗传探测技术在容量和准确性方面的快速提高,聚类分析在高吞吐量探测技术产生的大数据描述性建模(分割或分区)中发挥着更加重要的作用。但是,对于各种聚类分析算法的结果质量,尽管已经有了一些评价指标,在大多数实际研究项目中,其仍是由经验人员进行主观评判的,且往往仅特定于目标应用,并不具有普适性。为了选择数据集的最佳聚类方法及其参数,研究人员必须运行多个聚类算法并进行比较,而这种比较任务往往是十分苛刻和费力的。于是,为了解决这个问题,该章节的作者们开发了一款专门面向生物信息大数据的聚类结果可视化分析软件——XCluSim。该软件基本满足了以下4个设计目标:
(1)方便在不同层面上对多种聚类结果进行可伸缩的视觉比较;
(2)支持生成多种聚类结果;
(3)促进对各种聚类算法的特性及其结果中参数的理解;
(4)为不同类型的单个聚类结果提供有效的专用可视化。
XCluSim的优势
很遗憾的是,我们小组并没有找到获取XCluSim的途径,所以我们并没有亲身体验过XCluSim的强大功能。但是,通过对该章节内容的系统学习,我们已经深刻体会到该系统设计的严密性以及丰富的层次性,所以我们希望通过我们的简单介绍能让大家对这款软件有一个初步的了解。
(一)比较多个聚类结果
XCluSim部分工作界面如下:

显然,上图一共被分为五个部分:
(a)参数信息视图,向用户提供了所有聚类结果的参数概述。该视图被垂直划分为多个子视图,每个子视图对应单独的聚类算法。每个子视图内部又被划分为多个矩形区域,分别表示该聚类算法的不同参数。
(b)强定向布局概览,其通过物理距离直观地展示了各种聚类结果的相似性。在该视图中,每个聚类算法的结果都被表示为一个嵌入了饼状图的节点。其中,聚类结果越相似的节点靠得越近,而饼状图则向用户直观地展示了集群的数量和大小。
(c)树状图概览,其功能与强定向布局概览类似,都是向用户展示多种聚类结果的整体相似性。不同的是,该概览使用了更熟悉的可视化组件(即树状图),且相较于强定向布局概览,它节省了更多的空间而不会出现遮挡。
(d)增强版并行集视图,其更加详细地展示了多个聚类结果之间的一致性和不一致性。在并行集视图中,每一行水平的堆叠条都表示了一种聚类结果,而水平堆叠条中的每个条都表示聚类结果中的一个簇。除此之外,相邻两行堆叠条之间还存在许多宽度不一的线条,通过线条相连的簇具有某些相同的“稳定項”,而线条的宽度则表示了该稳定項的大小。用户可以通过调节视图底部的直方图来控制所显示稳定項的大小范围。
(e)列表视图,其通过分组模式和热图模式向用户具体地展示了所访问聚类结果的详细信息。在分组模式下,用户可以查看稳定组的数据;而在热图模式下,用户可以查看原始数据。
(二)查看单个聚类结果
XCluSim部分工作界面如下:
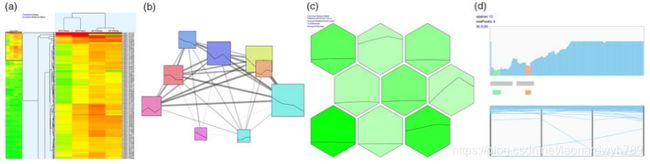
上图主要组成如下:
(a)树状图与热图的组合,用于分层显示聚类结果。
(b)簇级别的强定向布局,用于显示同一聚类结果中不同簇之间的相似性,其中的折线表示每个簇中所有成员对象的平均模式。
(c)SOM聚类【4】结果的通用蜂巢状可视化,其中每个六边形单元格都表示聚类结果中的一个簇。每个单元格的背景强度代表了相应簇的大小,而中间的折线图则显示了聚类成员的平均模式。
(d)可达性图及OPTICS平行坐标图,OPTICS会计算每个項的可达性距离。(略,这一块实在没看懂…)
(三)小结
通过对以上内容的学习,我们发现整个XCluSim的设计过程基本满足人们对于视觉信息的搜索认知:先概览,再缩放和过滤,以及按需获取详细信息。这种设计思路是十分值得我们进行借鉴和学习的。另外,XCluSim中对于颜色编码的使用也十分的巧妙。它使用了一种叫做Tree Colors【5】的颜色编码策略,它是为树状数据设计的,用于表示节点之间的相似性。其基本思路是父节点色调范围的一部分会通过递归的方式分配给它的子节点,这样就能使具有相同父节点的子节点具有相似的颜色。
拓展延伸
据文章所言,XCluSim可以很好地帮助生物学家挖掘某些基因的表达能力,具体案例包括了诸如阐明铁氧化酶在新型隐球菌中的作用等。在整个实验流程中,研究人员利用XCluSim对实验数据的多种聚类分析结果进行观察和比较,从中寻找某些比较稳定的簇群,而这些簇群往往代表着某些能够影响表达的基因序列。因为我们的小组成员都不是从事生物方向的,对于各种生物术语和实验流程的整体把握度还不够,所以就不在这里对具体实验实例进行过多阐述。
因为网上关于XCluSim这款软件的信息实在少得可怜,我们小组也没能够进行具体的尝试,无法对其作出具体的评估,所以只能在网上寻找一些我们认为能够帮助读者理解这篇文章内容的资料。具体内容如下:
(1)使用python演示如何通过聚类分析寻找共表达基因【6】。该文章详细地介绍了如何模拟数据集,如何使用K-means算法进行聚类分析,如何进行数据提取和可视化等步骤。
聚类模拟过程如下:
(来自 https://commons.wikimedia.org/wiki/File:Kmeans_animation.gif)

(2)一种基于语义的大型图像集可视化方法【7】。这是IEEE TRANSACTIONS ON VISUALIZATION AND COMPUTER GRAPHICS期刊于2018年发表的一篇文章。虽然该文章主要介绍的是一种针对大型图像集的可视化方法,但我们认为它的某些设计思想对XCluSim的发展有一定的帮助。该论文所描述的系统使用了星系比喻,其合理地使用各个独立的星系(如太阳系)来表示不同的簇,而星系中的恒星(如太阳)代表了该星系的特征,星系中的行星(如地球)和其他天体代表了簇中不同的对象个体。簇与簇之间的相似性可以用星系间的距离表示,簇的大小可以用星系的体积表示,簇中各个对象之间的相似性也可以通过空间距离来表示。相较于2D空间,星系比喻所使用的3D空间能够带给用户更加直观的感受和层次更加丰富的信息,我们相信这种方式能够更好地帮助XCluSim的用户理解和分析单个聚类算法的结果。
星系比喻的部分界面如下:

(3)基于MNIST数据集的无监督学习的一种简单应用——Encoder编码器【8】。其基本原理实际上就是将高维特征进行压缩降维编码,最后直观地在二维平面或三维空间中显示数据的聚类情况。
Tensorflow平台下的具体代码如下:
import tensorflow as tf
import matplotlib.pyplot as plt
from tensorflow.examples.tutorials.mnist import input_data
mnist = input_data.read_data_sets("MNIST_Labels_Images", one_hot=False)
# 设置训练超参数
learning_rate = 0.01 # 学习率
training_epochs = 10 # 训练次数
batch_size = 256 # 每次训练的样本数
display_step = 1 # 多少轮显示一次结果
n_input = 784 # 输入数据的维度
X = tf.placeholder("float", [None, n_input])
# 初始化权重和偏置
n_hidden_1 = 128
n_hidden_2 = 64
n_hidden_3 = 10
n_hidden_4 = 2
weights = {
'encoder_h1': tf.Variable(tf.truncated_normal([n_input, n_hidden_1], )),
'encoder_h2': tf.Variable(tf.truncated_normal([n_hidden_1, n_hidden_2], )),
'encoder_h3': tf.Variable(tf.truncated_normal([n_hidden_2, n_hidden_3], )),
'encoder_h4': tf.Variable(tf.truncated_normal([n_hidden_3, n_hidden_4], )),
'decoder_h1': tf.Variable(tf.truncated_normal([n_hidden_4, n_hidden_3], )),
'decoder_h2': tf.Variable(tf.truncated_normal([n_hidden_3, n_hidden_2], )),
'decoder_h3': tf.Variable(tf.truncated_normal([n_hidden_2, n_hidden_1], )),
'decoder_h4': tf.Variable(tf.truncated_normal([n_hidden_1, n_input], )),
}
biases = {
'encoder_b1': tf.Variable(tf.random_normal([n_hidden_1])),
'encoder_b2': tf.Variable(tf.random_normal([n_hidden_2])),
'encoder_b3': tf.Variable(tf.random_normal([n_hidden_3])),
'encoder_b4': tf.Variable(tf.random_normal([n_hidden_4])),
'decoder_b1': tf.Variable(tf.random_normal([n_hidden_3])),
'decoder_b2': tf.Variable(tf.random_normal([n_hidden_2])),
'decoder_b3': tf.Variable(tf.random_normal([n_hidden_1])),
'decoder_b4': tf.Variable(tf.random_normal([n_input])),
}
def encoder(x):
layer_1 = tf.nn.sigmoid(tf.add(tf.matmul(x, weights['encoder_h1']), biases['encoder_b1']))
layer_2 = tf.nn.sigmoid(tf.add(tf.matmul(layer_1, weights['encoder_h2']), biases['encoder_b2']))
layer_3 = tf.nn.sigmoid(tf.add(tf.matmul(layer_2, weights['encoder_h3']), biases['encoder_b3']))
# 为了便于编码层的输出,编码层随后一层不使用激活函数
layer_4 = tf.add(tf.matmul(layer_3, weights['encoder_h4']), biases['encoder_b4'])
return layer_4
def decoder(x):
layer_1 = tf.nn.sigmoid(tf.add(tf.matmul(x, weights['decoder_h1']), biases['decoder_b1']))
layer_2 = tf.nn.sigmoid(tf.add(tf.matmul(layer_1, weights['decoder_h2']), biases['decoder_b2']))
layer_3 = tf.nn.sigmoid(tf.add(tf.matmul(layer_2, weights['decoder_h3']), biases['decoder_b3']))
layer_4 = tf.nn.sigmoid(tf.add(tf.matmul(layer_3, weights['decoder_h4']), biases['decoder_b4']))
return layer_4
# 定义网络结构
encoder_op = encoder(X)
decoder_op = decoder(encoder_op)
y_pred = decoder_op
y_true = X
# 设置损失函数
cost = tf.reduce_mean(tf.pow(y_true - y_pred, 2))
optimizer = tf.train.AdamOptimizer(learning_rate).minimize(cost)
# 开始训练
with tf.Session() as sess:
init = tf.global_variables_initializer()
sess.run(init)
total_batch = int(mnist.train.num_examples / batch_size)
for epoch in range(training_epochs):
for i in range(total_batch):
batch_xs, batch_ys = mnist.train.next_batch(batch_size) # max(x) = 1, min(x) = 0
_, c = sess.run([optimizer, cost], feed_dict={X: batch_xs})
if epoch % display_step == 0:
print("Epoch:", '%04d' % (epoch + 1), "cost=", "{:.9f}".format(c))
print("Optimization Finished!")
encoder_result = sess.run(encoder_op, feed_dict={X: mnist.test.images})
plt.scatter(encoder_result[:, 0], encoder_result[:, 1], c=mnist.test.labels)
plt.colorbar()
plt.show()
聚类结果如下:

结语
总的来说,我们小组在这次学习过程中的收获还是蛮大的。虽然有些操作在没有实际尝试的情况下理解起来比较困难,同时对于一些生物方面的知识的缺乏也对我们造成了一定的困扰,但是这并不妨碍我们学习XCluSim这款软件所蕴含的设计思想和可视化技巧。我们相信在今后的学习生活中,我们一定会十分感谢这次学习经历的。
参考
【1】吴恩达 —— 无监督学习
https://blog.csdn.net/qq_29373285/article/details/82529333
【2】聚类分析的评价指标
https://blog.csdn.net/qq_36064669/article/details/82586582
【3】聚类分析和全基因组表达模式的展示
https://www.pnas.org/content/95/25/14863.short
【4】Self Organizing Maps(SOM):一种基于神经网络的聚类算法
https://www.cnblogs.com/sylvanas2012/p/5117056.html
【5】Tree Colors: Color Schemes for Tree-Structured Data
https://ieeexplore.ieee.org/abstract/document/6875961
【6】基因共表达聚类分析及可视化
https://blog.csdn.net/qazplm12_3/article/details/78904744
【7】A Semantic-based Method for Visualizing Large Image Collections
https://ieeexplore.ieee.org/abstract/document/8358974
【8】Tensorflow在MNIST中的应用
https://blog.csdn.net/hellozex/article/details/78540482