16/10/2019 一步步学会分析ATAC-seq
https://zhuanlan.zhihu.com/p/20702684 关于illumina 测序的原理
so, 我的测序结果是paired-end sequencing 并且是在不同的lanes上测序的。原始名称为:
larvae1_S1_L001_R1_001.fastq.gz ,
larvae1_S1_L001_R2_001.fastq.gz ,
larvae1_S1_L002_R1_001.fastq.gz ,
larvae1_S1_L002_R2_001.fastq.gz ,
larvae1_S1_L003_R1_001.fastq.gz ,
larvae1_S1_L003_R2_001.fastq.gz ,
larvae1_S1_L004_R1_001.fastq.gz ,
larvae1_S1_L004_R2_001.fastq.gz ,
#首先我需要把不同lane的sequencing 串联起来:
cat larvae1_S1_L001_R1_001.fastq.gz larvae1_S1_L002_R1_001.fastq.gz larvae1_S1_L003_R1_001.fastq.gz larvae1_S1_L004_R1_001.fastq.gz > larvae1_R1.fastq.gz
cat larvae1_S1_L001_R2_001.fastq.gz larvae1_S1_L002_R2_001.fastq.gz larvae1_S1_L003_R2_001.fastq.gz larvae1_S1_L004_R2_001.fastq.gz > larvae1_R2.fastq.gz
#接下来是质控: FastQC 继续在linux执行
module load fastqc
fastqc larvae1_R1.fastq.gz
fastqc larvae1_R2.fastq.gz
#接下来是去除adapter
有两种方法, cutadapt 是用于知道adapter序列的情况下; NGmerge 用于不知adapter序列的情况下, 并且只能用于paried-end测序的情况下。NGmerge 好像依赖于GCC,zlib OpenMP安装这些都是很头疼,我不知道怎么弄,直接用conda install zlib/GCC/OpenMP/NGmerge这样安装了。
module load NGmerge
NGmerge -1 larvae1_R1.fastq.gz -2 larvae1_R2.fastq.gz -o larvae1 -a
//
或者 NGmerge -a -1 larvae1_R1.fastq.gz -2 larvae1_R2.fastq.gz -o larvae1 -v
输出的文件为: larvae1_1.fastq.gz 和 larvae1_2.fastq.gz
接下来是和基因组匹配:
这里有一个比较重要的是,基因组序列必须建立索引。
module load bowtie2
bowtie2-build Amphimedon_queenslandica.Aqu1.dna.toplevel.fa Amphimedon_queenslandica.Aqu1.dna.toplevel
方法一:
bowtie2 -p 15 -x /opsin/u/huifang/ATAC-Seq/larvae1/ATAC-seq1017/Amphimedon_queenslandica.Aqu1.dna.toplevel -1 larvae1_1.fastq.gz -2 larvae1_2.fastq.gz -S larvae1.sam
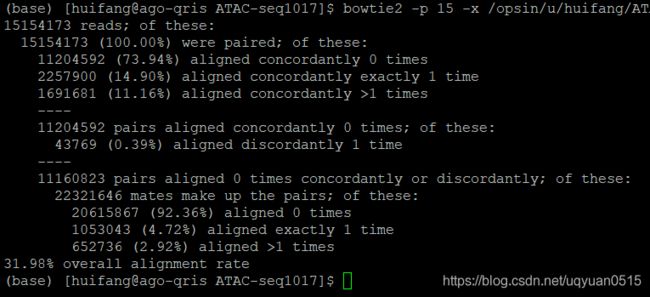
得到了一个很大的sam文件。
比对之后输出为sam文件,该文件包括了匹配信息。 然后Sam文件应该被压缩成bam文件,通过软件samtools 来实现。
grep -v "XS:i:" larvae1.sam |samtools view -bS - >larvae1-1.bam
samtools view -h larvae1-1.bam|less -SN
得到一个bam文件,bam是sam的压缩版文件,占空间少。
方法二:
以上也可以直接从bowtie2到samtools view and samtools sort, 输出bam 文件,
module load bowtie2 # if not already loaded module load samtools bowtie2 --very-sensitive -k 10 -xAqu2.1_Genes_mRNA-1larvae1_1.fastq.gz-2larvae1_2.fastq.gz\ | samtools view -u - \ | samtools sort -n -o larvae1 -
这一步可能会比较慢。查看bam文件用: samtools view -h larvae1.bam| less -SN
 但是我的bam文件都是这个样子,没有其他信息。。。。不知道是否正确。。。
但是我的bam文件都是这个样子,没有其他信息。。。。不知道是否正确。。。
总结,上述两种方法我都试了,结果还是先转换为sam文件然后转换为bam文件比较靠谱。第二个一步法不行。。。得到的数据不全,虽然不知道为什么。。。
samtools sort -n larvae1.bam -o larvae1_1.bam
新的bam文件为queryname 比对后的bam文件,要不然没法用Genrich.
#接下来是peak-calling
Harvard Uni 推荐使用新的软件Genrich 可以从Github下载。 好处是可以出去线粒体reads, PCR duplicates, 用于多重mapping分析,以及分析多个replicates. 适用于ATAC-seq的线性匹配解释。 Harvard对比了之前的MACS2之后,推荐使用 Genrich.
软件可以从Github 下载,conda install Genrich 安装
第一次尝试:
Genrich -t larvae1_1.bam -o larvae1.narrowPeak -f larvae1.log -r -x -q 0.05 0a 20.0 -v -e chrM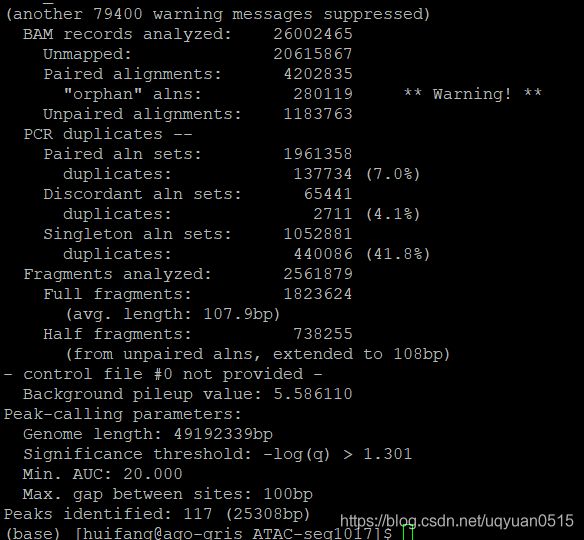
Genrich -P -f larvae1.log -o larvae1_p01_a200.narrowPeak -p 0.01 -a 200 -v
这步出来了540多个peak.
第二次尝试:
Genrich -t larvae1_1.bam -o larvae1.bed -j -y -e chrM -v
出来了835个peak。
#结果我觉得得到的peak都太少了,我还是用了MACS14,去callpeak.
macs14 -t larvae1.bam -f BAM -n larvae1019 -B
结果呢,这个跑了大概14个小时,得到了大概一万六千个peak. 以及excel表格,和peak.bed 文件,以及R脚本。通过运行得到的R脚本,可以得到peak Model 的图谱,然后也可以根据这个excel表格得到Peak分布的柱状图。
接下来的步骤都是在R里进行的,可根据chipseeker 的document 来操作。 主要使用了peaks.bed这个文件
#接下来是注释:使用chip-seeker 在R里运行,chipseeker 是一个R包
使用ChIPseeker需要准备两个文件:一个就是要注释的peaks的文件,需满足BED格式。另一个就是注释参考文件,即需要一个包含注释信息的TxDb对象。我从上面的分析得到了peak.bed文件。但是没有TxDb 文件啊,使用方法是从Ensembl下载gff格式的基因组文件,然后使用GenomicFeatures 包来制作TxDb.
require(GenomicFeatures)
spombe<-makeTxDbFromGFF("Amphimedon_queenslandica.Aqu1.45.gff3")
这样就可以了。
使用chipseeker, 第一步,安装相关的包。这里必须记一下chipseeker包原创者的博客地址,这样方便以后的学习/复习/解疑//https://guangchuangyu.github.io/cn/tags/chipseeker/
library(S4Vectors)
library(clusterProfiler)
library(GenomicFeatures)
library(ggplot2)
library(RMariaDB)
library(GenomicRanges)
library(ChIPseeker)
library(ChIPpeakAnno)
library(annotatr)
##################start at there################################useEnsembl to get a TxDb
require(GenomicFeatures)
txdb<-makeTxDbFromGFF("Amphimedon_queenslandica.Aqu1.45.gff3")
gff文件下载与ensemble, 是基因组的注释文件。
peak <- readPeakFile("larvae1023_peaks.bed")
peak
covplot(file, weightCol="V5")
dev.off()##mapping peaks in chr
promoter<-getPromoters(TxDb = txdb, upstream = 3000, downstream = 3000)
tagMatrix<- getTagMatrix(file, window=promoter)
###plot the heatmap of tagMatrix
tagHeatmap(tagMatrix,xlim = c(-3000,3000), color = "red")###plot the heatmap of peaks align to flank sequences of TSS
peakHeatmap("larvae1023_peaks.bed",
weightCol = NULL,
TxDb = txdb,
upstream = 3000, downstream = 3000,
xlab = "", ylab = "", title = NULL,
color = "red",
verbose = TRUE)
dev.off()
##这两个热图是一样的
###########map two TSS distribution pictures
plotAvgProf(tagMatrix, xlim=c(-3000, 3000),
xlab = "Genomic Region (5'->3')",
ylab = "Read Count Frequency",
conf=0.95, resample=1000)plotAvgProf(tagMatrix, xlim=c(-3000, 3000),
xlab = "Genomic Region (5'->3')",
ylab = "Read Count Frequency")
dev.off()#得到的一个是带阴影的,一个是不带的 TSS 分布图
#接下来是peak的注释
peakAnno <-annotatePeak("larvae1023_peaks.bed",
tssRegion = c(-3000, 3000),
TxDb = txdb,
level = "transcript",
assignGenomicAnnotation = TRUE,
genomicAnnotationPriority = c("Promoter", "5UTR", "3UTR", "Exon", "Intron", "Downstream", "Intergenic"),
annoDb = NULL,
addFlankGeneInfo = FALSE,
flankDistance = 5000,
sameStrand = FALSE,
ignoreOverlap = FALSE,
ignoreUpstream = FALSE,
ignoreDownstream = FALSE,
overlap = "TSS",
verbose = TRUE)#peak的注释
#接下来是peak注释的可视化
plotAnnoBar(peakAnno)
plotAnnoPie(peakAnno)
vennpie(peakAnno)
upsetplot(peakAnno)
upsetplot(peakAnno, vennpie=TRUE)
#Visualize distribution of TF-binding loci relative to TSS
plotDisToTSS(peakAnno, title="Distribution of transcription factor-binding loci\nrelative to TSS)
#接下来是功能富集分析:
Go /KEGG 富集分析
############GO enrich and kegg enrich################################
require(AnnotationHub)
hub <- AnnotationHub()
query(hub, "Amphimedon queenslandica")
aqsponge<-hub[["AH66597"]] #aqsponge.OrgDb <- hub[["AH10455"]]
length(keys(aqsponge))[1]
columns(aqsponge)require(clusterProfiler)
#改变ID格式
bitr(keys(aqsponge)[1], 'ENTREZID', c("REFSEQ", "GO", "ONTOLOGY"), aqsponge)
sample_genes <- keys(aqsponge)[1:100]
head(sample_genes)# still need the "peak.bed"
txdb<- makeTxDbFromGFF("Amphimedon_queenslandica.Aqu1.45.gff3")
peak<-readPeakFile("larvae1023_peaks.bed")
peakAnno <-annotatePeak("larvae1023_peaks.bed",
assignGenomicAnnotation = TRUE,
tssRegion = c(-3000, 3000),
TxDb=txdb,addFlankGeneInfo = TRUE,
flankDistance = 5000)
library(GenomicRanges)library(dplyr)
as.GRanges(peakAnno) %>% head(3)###start GO enrich, we need geneID
gene=as.data.frame(peakAnno)$geneId
###Error: operator not defined for this S4 class
kk<- enrichGO(gene =sample_genes, OrgDb= aqsponge,
pvalueCutoff = 0.05,
qvalueCutoff = 0.05,
ont="all",
readable = T)write.table(kk, file = "GO.txt", sep="\t", quote = F, row.names = F)
两种图表显示
#####bar
pdf(file="barplot.pdf", width=12, height=7)
barplot(kk, drop=TRUE, showCategory=10, split="ONTOLOGY")+ facet_grid(ONTOLOGY~., scale='free')
####bubble
pdf(file="bubble.pdf",width = 12,height = 7)
dotplot(kk,showCategory = 10,split="ONTOLOGY") + facet_grid(ONTOLOGY~., scale='free')
dev.off()#同样的进行KEGG分析
gene =as.data.frame(peakAnno)$geneID
kk<-enrichKEGG(gene = sample_genes, organism="", pvalueCutoff = 0.05, qvalueCutoff = 0.05)
write.table(kk, file="KEGG.txt", sep="\t", quote=F,row.names = F )###bar
pdf("barplotkegg.pdf", width=12, height=7)
barplot(kk, drop=TRUE, showCategory =20)
dev.off()
###bubble
pdf("bubblekegg.pdf", width = 12, height=7)
dotplot(kk, showCategory =20)
dev.off()
#接下来就是查找mtif 了。
首先要将peaks.bed文件转换为fasta文件
在Linux下进行的: 用了bedtools 工具
$ bedtools getfasta -fi Amphimedon_queenslandica.Aqu1.dna.toplevel.fa - bed larvae1023_peaks.bed -fo larvae1023_peaks.fa.out
这就转换成了fasta文件,然后将文件输入到 MEME suit 网站上进行分析, 选择了meme-chip工具