注:安装环境:Ubuntu 16.04 LTS,LAMMPS-14May16,MPICH3.2,FFTW3.3.4,VMD1.9.3
LAMMPS(Large-scale Atomic/Molecular Massively Parallel Simulator)是一款经典的开源分子动力学软件,对于求解凝聚态材料、软物质等体系效率很高,在材料模拟领域有很广泛的应用。本文主要介绍LAMMPS的单机版、并行版以及超算中心集群上的安装。
1 LAMMPS单机版安装
解压后 cd lammps-14May16/src
打开Makefile.serial,目录为lammps-14May16/src/MAKE/
下面的几行的意思是为计算机搭建一个虚拟的MPI库STUBS
MPI_INC = -I../STUBS
MPI_PATH = -L../STUBS
MPI_LIB = -lmpi_stubs单机版安装很简单,在src目录下输入命令make serial,可以发现生成了lmp_serial可执行文件,然后进入example/shear目录测试一下,在in.shear文件中寻找下面这一行:
#dump 1 all atom 100 dump.shear 删掉#后运行../../src/lmp_serial < in.shear,将会生成dump.shear文件,之后将会用它来进行可视化。
2 LAMMPS并行版安装
首先下载安装FFTW3和MPICH3,后者可以用OpenMPI替代。
安装FFTW3.3.4
默认FFTW会安装到/usr/local/下,但是安装目录可以改变./configure --prefix=/home/foo/fftw
chmod +x configure
./configure
make
sudo make install安装MPICH
进入解压后的文件夹后
mkdir /home/foo/mpich-install
./configure --prefix=/home/foo/mpich-install 2>&1 | tee c.txt
make 2>&1 | tee m.txt
sudo make install 2>&1 | tee mi.txt安装好了mpich以后需要设置环境变量,在用户目录下输入gedit .bashrc,在最后添加下面两行,保存后输入命令source .bashrc即可
PATH=/home/foo/mpich-install/bin:$PATH ; export PATH
export LD_LIBRARY_PATH=$PATH:/home/foo/mpich-install/lib输入which mpicxx,看看返回的是不是正确的路径,如果正确的话说明安装成功。
现在必要的软件都安装好了,假设你之前安装过单机版的LAMMPS,首先进入lammps-14May16/src目录清理一下sudo make clean-all。如果有特殊需求,那么在安装lammps可执行文件之前要先安装一些额外的包,如在正式编译之前输入这样的命令make yes-class2 yes-rigid yes-shock,即首先要安装class2、rigid、shock包。这些包的安装比较简单,但还有一些包的安装需要额外的库,如果在实际科研中用到,参考Manual安装即可。
编译LAMMPS
注:编译的时候Makefile里的编译器选项如果设置成mpicxx的话,后面的MPI相关选项就可以不必设置
下面是简便的编译方案——在MAKE/OPTIONS目录里面寻找Makefile.g++_mpich和Makefile.fftw这两个Makefile,把后者设置fftw的部分整体复制到Makefile.g++_mpich里的对应位置(如果前面修改了fftw的安装目录,则要做必要的修改),将这个文件复制到MAKE/MINE里面,修改文件名为Makefile.foo
回到src目录,命令行输入sudo make foo -j 4,注意这里的foo即为Makefile.xxx的后缀(我用了4个核来编译,可以在make后紧接着加-j N来表示使用N个核编译,如果顺利的话,可以生成lmp_foo可执行文件
将LAMMPS添加到环境变量
不想在以后调用lammps求解时都得输入绝对路径,那就把它加到环境变量里吧(参考上面的添加方法):
PATH=/home/foo/lammps-14May16/src:$PATH ; export PATH测试一下
cd ../examples/shear/在in.shear文件中寻找下面这一行,并将前面的#删掉:
#dump 1 all atom 100 dump.shear运行mpiexec -n 4 lmp_foo < in.shear,顺利的话可以生成dump.shear文件。
其他莫名其妙的错误
大多数错误都源自于少了某些依赖包(看到那些红色的fatal error尤其可能),使用sudo apt-get + Baidu就可以解决大部分问题,因此不必慌乱。
3 LAMMPS集群安装
在一般的超算平台上编译,需要ssh到编译节点,例如ssh gs0110,然后在用户节点下编译fftw
如果计算中心节点有openmpi并行环境,修改Makefile.g++_openmpi文件,把fftw3的标识路径按照Makefile.fftw的形式修改为自己安装的路径。最后修改Makefile.g++_openmpi文件名为Makefile.foo
进入src文件夹,make foo -j 12
4 使用VMD可视化
计算完了,看着命令行飞速的闪过一行行的数据,很过瘾吧:P。可是计算完了以后呢?LAMMPS到底干了些什么?这就需要进行可视化,把计算结果用图片或者动画的形式形象的展现在我们的眼前,下面我就用前面产生的dump文件,用VMD来进行可视化操作。
进入VMD网站,提示还要注册,没关系。下载一个对应于自己计算机的压缩包,我下载的是vmd-1.9.3.bin.LINUXAMD64-CUDA8-OptiX4-OSPRay111p1.opengl.tar.gz。进入文件夹后,注入命令./configure会提示你输入在后面加上LINUXAMD64(或别的类似的),因此安装流程如下:
cd vmd-1.9.3
./configure LINUXAMD64
cd src
sudo make install安装成功以后直接在命令行里输入vmd就可以将程序调出来,选择New Molecule,然后导入我们之前计算得到的dump.shear文件,记得在Determine file type一栏选择LAMMPS trajectory选项。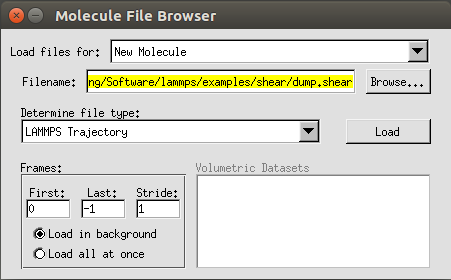
导入以后,可以得到这样的构型,拖动main对话框下面的滑动条就可以看到体系的剪切行为,大功告成!
5 MD相关资源推荐
对刚刚接触这一个领域的初学者而言,一些指引是很有必要的。我就把我觉得有用的资源贴在这里,以后也会经常更新,供需要的同学参考。
祝大家学习愉快,以后的科研生活步步顺利!
软件:
- VMD:不用多说了吧,很强大的一款后处理软件。
- Atomeye:MIT的一位博士自己写的,通过一些指令来实现可视化操作,使用起来很方便。
- Avogadro:界面很友好,开源的,在Ubuntu下需要安装各种各样的依赖软件,我没弄成功,所以在Windows里使用。我感觉和Materials Studio有点类似,直接能够建立分子甚至DNA双螺旋结构。
- EMC:主要用来建立随机结构。
- Packmol: 命令很简单,用于建立分子随机排布效果挺好的。
- moltemplate:很强大,我用的很多。可以按照自己的想法来建立模型,没有图形界面,主要通过代码式的操作,因此需要和VMD等联合起来使用。
- OVITO:全平台的可视化软件,Windows和Ubuntu中都可以使用,操作简便。
资料:
- 用matlab写一个100行的分子动力学模拟程序:大神写的MATLAB程序,可以参考着理解MD算法。
- 《计算化学——从理论到分子模拟》 陈敏伯 著
- 《分子模拟的理论与实践》 陈正隆 著