转录组软件安装及分析流程(Hisat2-Stringtie-Ballgown)
替换镜像源,提高下载速度
为了提高下载速度,我们需要替换/etc/apt/source.list中默认镜像源。方法参考自中国科学技术大学开源镜像站
备份
cd /etc/apt/
sudo cp source.list source.list.bk
替换
sudo sed -i ‘s/http/https/g’ sources.list
sudo sed -i ‘s/archive.ubuntu.com/mirrors.ustc.edu.cn/g’ sources.list
sudo sed -i ‘s/security.ubuntu.com/mirrors.ustc.edu.cn/g’ sources.list
更新
sudo apt-get update
sudo apt-get upgrade
下载数据
download singcell Rseq data SRR
for ele in {511..519}
do
echo “https://sra-download.ncbi.nlm.nih.gov/srapub/SRR2089$ele” >> download.txt
done
wget -ci download.txt
sratookit下载安装
功能: 下载,操作,验证NCBI SRA中二代测序数据
网址:https://trace.ncbi.nlm.nih.gov/Traces/sra/sra.cgi?view=software
步骤:
cd src
wget https://ftp-trace.ncbi.nlm.nih.gov/sra/sdk/2.8.2-1/sratoolkit.2.8.2-1-ubuntu64.tar.gz
tar -zxvf sratoolkit.2.8.2-1-ubuntu64.tar.gz
mv sratoolkit.2.8.2-1-ubuntu64 ~/biosoft
加入环境变量
echo ‘PATH=$PATH:~/biosoft/sratoolkit.2.8.2-1-ubuntu64/bin’ >> ~/.bashrc
source ~/.bashrc
测试
prefetch -v
prefetch -c SRR2413322
SRA转换为fastq格式
for i in {511..519}
do
echo “fastq-dump –split-3 SRR2089$i -O ../fastq” >> ../fastq/fastq.sh
done
运行fastq.sh
SAMtools下载安装
SAM(sequence Alignment/mapping)数据格式是目前高通量测序中存放比对数据的标准格式,当然他可以用于存放未比对的数据。目前处理SAM格式的工具主要是SAMTools,这是Heng Li大神写的。SAMTools的主要功能如下:
view: BAM-SAM/SAM-BAM 转换和提取部分比对
sort: 比对排序
merge: 聚合多个排序比对
index: 索引排序比对
faidx: 建立FASTA索引,提取部分序列
tview: 文本格式查看序列
pileup: 产生基于位置的结果和 consensus/indel calling
下载 wget https://nchc.dl.sourceforge.net/project/samtools/samtools/1.6/samtools-1.6.tar.bz2
解压 tar jxvf samtools-1.6.tar.bz2
添加到环境变量
echo ‘PATH=$PATH:~/biosoft/samtools-1.6’ >> ~/.bashrc
source ~/.bashrc
Hisat2创建基因组索引
人和小鼠的index一般都有现成的,建议大家下载现成的
http://ccb.jhu.edu/software/hisat2/index.shtml 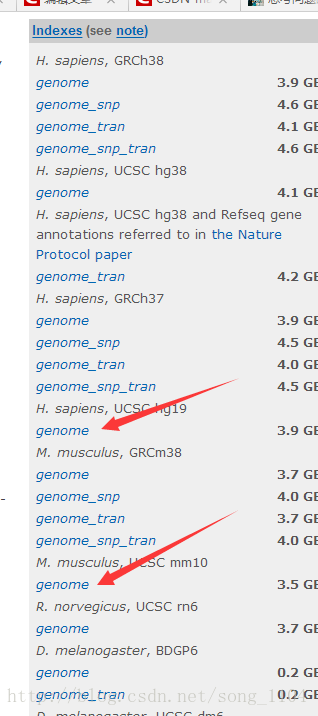
wget ftp://ftp.ccb.jhu.edu/pub/infphilo/hisat2/data/hg19.tar.gz
tar -zxvf hg19.tar.gz
而猪的基因组没有现成的Index,需要我们手动创建
HISAT2提供两个Python脚本将GTF文件转换成hisat2-build能使用的文件:
extract_exons.py Sus_scrofa.Sscrofa11.1.90.chr.gtf > genome.exon
extract_splice_sites.py Sus_scrofa.Sscrofa11.1.90.chr.gtf > genome.ss
最后创建Index
hisat2-build –ss genome.ss –exon genome.exon Sus_scrofa.Sscrofa11.1.dna.toplevel.fa Sus_tran 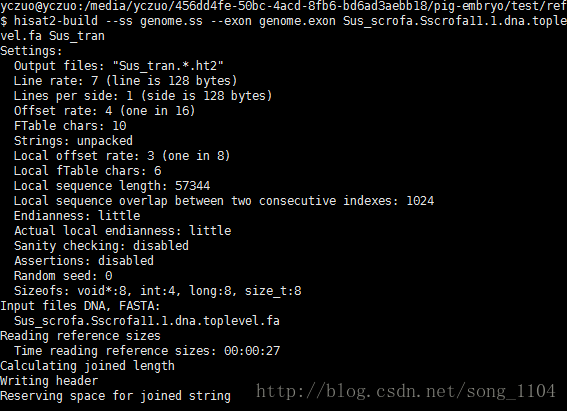
Hisat2比对
将RNA-seq的测序reads使用hisat2比对
hisat2 -p 8 –dta -x ./ref/Sus_tran/Sus_tran -1 ./fastq/Blast_1.clean.fq -2 ./fastq/Blast_2.clean.fq -S ./hisat2-out/Blast.sam 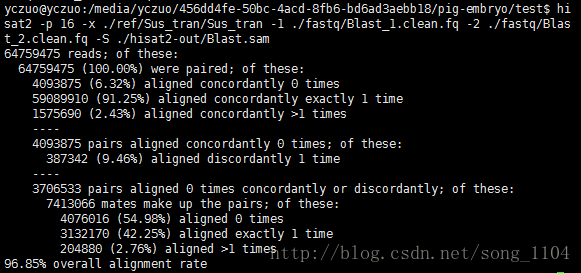
比对率达到了96.85,大功告成。
HTSeq安装
使用pip直接下载:
pip install HTSeq
如果失败了,下载依赖包:
pip install ‘matplotlib>=1.4’
pip install Cython
pip install ‘pysam>=0.9’
pip install HTSeq
如果还失败,使用 setup.py
wget https://github.com/simon-anders/htseq/archive/master.zip
解压进入该目录,输入 python setup.py install –user
程序的路径 .local/bin ![]()
添加到环境变量就可以了或者
/home/yczuo/.local/bin/htseq-count Blast.sam /home/yczuo/ref/Sus.gtf
htseq-count 计数
将sam文件转换为bam文件
samtools view -S ./hisat2-out/Blast.sam -b > ./BAM/Blast.bam
bam文件排序#因为是双端测序,必须对bam文件排序
samtools sort -n ./BAM/Blast.bam ./BAM/Blast_sort.bam
samtools view -h ./BAM/Blast_sort.bam > ./SAM/Blast_sort.sam
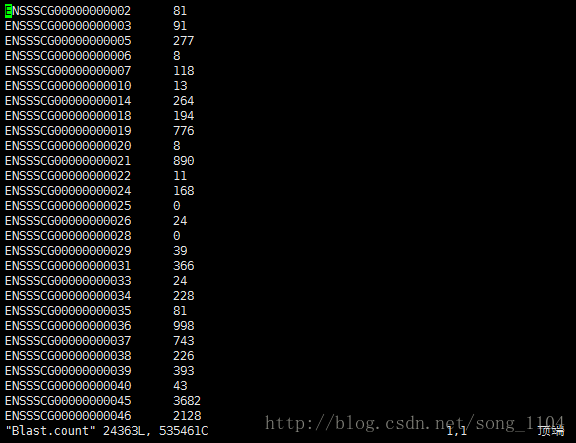
htseq-count -s no ./SAM/Blast_sort.sam genes.gtf > ./reads count/Blast.count
写个循环
for ele in Blast ICM Morula Oocyte P1_cell P2_cell P4_cell P8_cell PFF TE
do
echo -e “samtools view -S ./hisat2-out/ele.sam−b>./BAM/ele.bam\nsamtools sort -n ./BAM/ele.bam./BAM/ele.sort\nsamtools view -h ./BAM/ele.sort.bam>./SAM/ele.sort.sam\nhtseq-count -s no ./SAM/ele.sort.sam./ref/Susscrofa.Sscrofa11.1.90.chr.gtf>./count/ele.count\n” >> htseq.sh
done
chmod 777 ./htseq.sh
nohup ./htseq.sh > htseq.log 2>&1 &
查看任务
jobs -l
ps -ef |grep htseq 
结果输出count值 
stringtie安装
wget http://ccb.jhu.edu/software/stringtie/dl/stringtie-1.3.3b.Linux_x86_64.tar.gz
解压 tar zxvf stringtie-1.3.3b.Linux_x86_64.tar.gz
添加到环境变量
echo ‘PATH=$PATH:~/biosoft/stringtie-1.3.3b.Linux_x86_64’ >> ~/.bashrc
source ~/.bashrc
stringtie转录本处理
1、 stringtie组装转录本(首先将sam文件转换为bam文件,并排序;然后对每个样本进行转录本组装)
for ele in Blast ICM Morula Oocyte P1_cell P2_cell P4_cell P8_cell PFF TE
do
echo -e “samtools view -S ele.sam−b>ele.bam\nsamtools sort -@ 8 ele.bamele.sorted\nstringtie -p 8 -G Sus.gtf -o ele.gtfele.sorted.bam” >> out.sh
done 
2 、stringtie合并转录本(将所有样本的转录本进行合并)
stringtie –merge -p 8 -G Sus.gtf -o stringtie_merged.gtf mergelist.txt #mergelist.txt是自己创建的
for ele in Blast ICM Morula Oocyte P1_cell P2_cell P4_cell P8_cell PFF TE
do
echo -e “./$ele.gtf” >> mergelist.txt
done 
3、stringtie评估表达量(计算表达量并且为Ballgown包提供输入文件)
for ele in Blast ICM Morula Oocyte P1_cell P2_cell P4_cell P8_cell PFF TE
do
echo -e “stringtie -p 8 -G stringtie_merged.gtf -e -B -o ballgown/ele/ele.gtf $ele.sorted.bam” >> out2.sh
done 
在-B 指定的文件夹下生成特定的文件
e2t.ctab e_data.ctab i2t.ctab i_data.ctab t_data.ctab
e即外显子、i即内含子、t转录本;e2t即外显子和转录本间的关系,i2t即内含子和转录本间的关系,t_data即转录本的数据 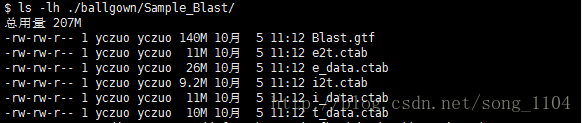
Ballgown表达量分析
1、 Ballgown的安装
source(“http://bioconductor.org/biocLite.R“)
biocLite(“ballgown”)
2、文件准备与分析
将数据的分组信息写入一个csv文件,此处phenodata.csv文件 
3、运行R脚本,分析
Rscript expr.R
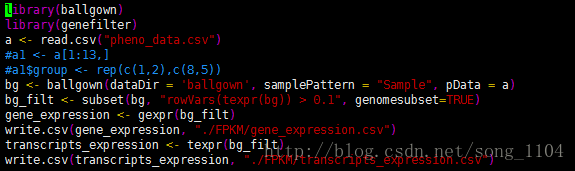
library(ballgown)
library(genefilter)
a <- read.csv(“pheno_data.csv”)
bg <- ballgown(dataDir = ‘ballgown’, samplePattern = “Sample”, pData = a)
bg_filt <- subset(bg, “rowVars(texpr(bg)) > 0.1”, genomesubset=TRUE)
gene_expression <- gexpr(bg_filt)
write.csv(gene_expression, “./FPKM/gene_expression.csv”)
transcripts_expression <- texpr(bg_filt)
write.csv(transcripts_expression, “./FPKM/transcripts_expression.csv”)